In-Depth Analysis of Critical Quality Attributes (CQAs) for AAV
Release time:2025-12-05 15:59:55
In the field of gene therapy, the quality of adeno-associated virus (AAV) vectors directly determines the safety and efficacy of treatments, with Critical Quality Attributes (CQAs) forming the core framework of the quality control system. The CQAs of AAV vectors must comprehensively cover five major dimensions: identity, quantity & potency, purity, impurities, and safety.
These factors influence transduction efficiency, biodistribution, and immunogenicity; therefore, evaluating them is essential for therapeutic success. Routine characterization of these attributes provides valuable insights into product consistency and safety.
This article integrates analytical techniques, quality control standards, and application scenarios to dissect these five dimensions in depth, offering a comprehensive view of the core objectives and technical requirements for AAV quality control.
I. Identity: Confirming the “Correct Vector” and Eliminating Mismatch Risks
Identity testing is the first checkpoint in AAV quality control. Its core objective is to verify the authenticity of the vector — ensuring that the produced AAV matches the intended design, without serotype mismatch, sequence mutation, or cross-contamination. This prevents “identity errors” that could lead to targeting failure or increased immunogenicity.
According to ICH Q6B guidelines, AAV identity must be validated across two dimensions: protein-level and nucleic acid–level characterization. The quality standards and analytical methods are outlined below.
1. Capsid Protein Identification: Verifying the Vector’s “External Features”
The AAV capsid consists of VP1, VP2, and VP3 in a 1:1:10 ratio. Different serotypes (e.g., AAV2, AAV5, AAV8, AAV9) show distinct molecular weights and amino acid sequences, which serve as the primary basis for protein-level identification.
(1) Common analytical methods: SDS-PAGE, CE-SDS
◦ SDS-PAGE with silver staining: This method denatures proteins and separates VP1/VP2/VP3 based on molecular weight. After silver staining, three distinct bands should appear, with molecular weights matching the target serotype (e.g., for AAV9: VP1 ~87 kDa, VP2 ~73 kDa, VP3 ~62 kDa). Although easy to operate, SDS-PAGE has limited resolution and cannot reliably distinguish closely related serotypes. It is suitable for preliminary serotype screening rather than precise identification.
◦ CE-SDS (Capillary Electrophoresis–SDS): Using a capillary as the separation channel, CE-SDS enables high-voltage, high-resolution protein separation, combined with UV/fluorescence detection. It allows accurate quantification of the VP1/VP2/VP3 ratio (must align with the theoretical 1:1:10 range, deviation ≤10%). Its superior resolution enables discrimination between closely related serotypes such as AAV2 and AAV3. CE-SDS is currently the FDA/NMPA recommended first-line method for capsid identification.
(2) Quality requirements:
Must rule out capsid contamination from other serotypes.For engineered capsids (e.g., AAV-PHP.B), peptide mapping (HPLC-MS) must confirm alignment with the designed amino acid sequence, ensuring that no mutations compromise function.
2. Feature Sequence Identification: Verifying the Vector’s “Internal Core”
The functionality of an AAV vector relies on its feature sequences — including the inverted terminal repeats (ITRs), therapeutic gene (GOI), promoter, and other regulatory elements. Any deletion or mutation may render the vector ineffective. Thus, nucleic acid–level identification focuses on validating the integrity and correctness of these essential sequences.
(1) Common analytical methods: PCR, sequencing, Southern blot
◦ PCR (Polymerase Chain Reaction): Primers targeting ITRs or GOI-specific regions amplify fragments whose band sizes (after gel electrophoresis) must match expected values, confirming the presence of key sequences. ◦ Gene sequencing: Sanger sequencing (first-generation): Precisely confirms base sequences in crucial regions such as coding areas, ensuring no point mutations, insertions, or deletions. NGS (next-generation sequencing): Extracted AAV genomes are sequenced in bulk to obtain complete genomic coverage, enabling detection of SNVs, insertions, and deletions. Third-generation sequencing (e.g., PacBio SMRT): Capable of reading the full ~4.7 kb genome in a single pass and overcoming sequencing interruptions caused by ITR secondary structures (e.g., hairpin loops). Particularly valuable for validating complex vectors such as dsAAV or scAAV. ◦ Southern blot: Using labeled nucleic acid probes, Southern blot detects specific DNA sequences through hybridization (base-pairing), enabling analysis of genome structure, copy number, rearrangement, or deletions.
(2) Quality requirements:
Sequencing coverage must reach 100%.Base-calling accuracy must be ≥99.99%.Must exclude contamination with wild-type AAV sequences (wild-type AAV contains rep/cap, which may trigger immune responses).
II. Quantity & Potency: Ensuring Precise Dosing and Balancing Efficacy with Safety
The quantity of an AAV vector reflects the total number of particles, while potency reflects the number of functionally active vectors. Together, they determine the clinical dosage — insufficient dosage leads to therapeutic failure, whereas excessive dosage may trigger immune responses (e.g., antibody responses against capsid proteins).
Therefore, accurate quantification of quantity and potency is a core metric in AAV quality control, requiring simultaneous measurement of genomic titer, capsid titer, infectious titer, and biological activity.
1. Genomic Titer: Quantifying the Total Number of Vectors Carrying the Therapeutic Gene
Genomic titer (GT) refers to the number of AAV particles containing a complete genome per milliliter of sample, expressed as genome copies per mL (GC/mL) or vector genomes per mL (vg/mL).
(1) Common Quantification Methods: Fluorescent Dye Assay, qPCR, and ddPCR
• Fluorescent Dye Assay
Double-stranded DNA (dsDNA) fluorescent dyes bind specifically to dsDNA and generate strong fluorescence at 480 nm excitation, with emission detectable at 520 nm. Within an appropriate concentration range and dye excess, fluorescence intensity is proportional to DNA concentration. The DNA content in the test sample is calculated based on the measured fluorescence signal.
• qPCR (Quantitative Real-Time PCR)
Using primers targeting ITRs or GOI sequences, qPCR amplifies DNA with fluorescence-based quantification. Genomic titer is calculated by comparison with a standard curve. This method is fast and cost-effective but may be affected by free plasmids or incomplete genomes. Therefore, DNase I pretreatment is required to degrade unpackaged DNA before testing.
• ddPCR (Digital PCR)
The sample is diluted to single-molecule levels and partitioned into microdroplets, enabling absolute quantification without standard curve calibration. ddPCR shows superior resistance to inhibitors and high reproducibility. It has been recommended by regulatory agencies as the gold standard for determining genomic titer.
Method
Key Advantages
Main Limitations
Applicable Scenarios
Fluorescent staining
Easy to operate; low cost; detects total DNA
Cannot distinguish specific vs. non-specific DNA; susceptible to contamination from residual DNA in reagents
Rapid screening and preliminary control of total DNA residues during production
qPCR
Low cost; fast; high sensitivity; widely available instrumentation
Requires standard curves; susceptible to PCR inhibitors and non-target DNA interference
Rapid and sensitive quantification of target sequences during R&D and pilot production
ddPCR
Absolute quantification (no standard curve needed); strong resistance to inhibitors; high precision and repeatability
Higher equipment and consumable costs; relatively lower throughput
Accurate quantification for critical stages such as clinical submissions and commercial batch release
2. Capsid Titer: Quantifying the Total Number of Capsid Particles
Capsid titer (CT) refers to the total number of AAV capsid particles per milliliter of sample—regardless of whether they contain a genome. This metric is essential for assessing batch-to-batch consistency and purification efficiency. Capsid titer is expressed as virus particles per milliliter (vp/mL). Comparing capsid titer with genomic titer helps determine the full-to-empty capsid ratio, a key indicator of vector quality.
(1) Common Analytical Methods: ELISA, HPLC, AUC, BLI
• ELISA (Enzyme-Linked Immunosorbent Assay): Uses serotype-specific capsid antibodies for immunodetection. It offers high specificity and good standardization but requires serotype-matched antibodies and traceable reference standards.
• HPLC (High-Performance Liquid Chromatography): Detects capsid particles based on intrinsic physicochemical properties of capsid proteins. It provides rapid analysis and can determine both capsid titer and the ratio of empty to full particles in a single run.
• AUC (Analytical Ultracentrifugation): Under strong centrifugal force, particles with different sedimentation coefficients—such as full capsids, empty capsids, and aggregates—sediment at different rates. The optical detection system continuously monitors sedimentation to generate a sedimentation profile, allowing precise quantification of full capsids, empty capsids, partially filled capsids, and aggregates.
• BLI (Bio-Layer Interferometry): Capsid-specific antibodies are immobilized on the sensor surface, and real-time changes in optical interference signals are used to quantify capsid concentration. BLI requires no wash steps, offers fast turnaround (≈30 minutes), and supports multi-sample analysis. However, it is sensitive to particle aggregates, which may cause artificially elevated signals; therefore, samples should be filtered through a 0.22 μm membrane to remove large aggregates before testing.
Method
Key Advantages
Main Limitations
Applicable Scenarios
ELISA
High specificity; standardized workflow, easy to implement in QC labs
Requires serotype-matched antibodies; reference standards must be calibrated using methods such as AUC
Commercial-scale production and QC release of common serotypes (e.g., AAV2/8/9)
HPLC
Fast analysis; provides capsid titer and empty/full ratio in one run
Requires extensive method development; resolution generally lower than AUC
Rapid component analysis during process development and optimization
AUC
Extremely high resolution; precisely distinguishes and quantifies full, empty, partially filled capsids, and aggregates; regarded as the benchmark method
Expensive instrument; long turnaround time; complex operation requiring skilled personnel
Authoritative method for reference standard characterization and resolving critical disputes
BLI
Very short analysis time (≈30 min); real-time monitoring; supports parallel multi-sample analysis
Highly sensitive to particle aggregation; requires strict sample pretreatment; antibody immobilization is a technical challenge
Process optimization and rapid batch screening during pilot-scale manufacturing
3. Biological Activity: Verifying the Functional Efficacy of the Therapeutic Gene
Biological activity is a core functional metric for AAV vectors. It evaluates whether the therapeutic gene product exhibits the expected biological effect (e.g., enzymatic activity, signaling pathway modulation) at the cellular or animal level. This ensures that mutations in the gene sequence or promoter failure do not lead to functional loss.
3.1 Infectious Titer (IT)
Infectious titer refers to the number of AAV particles per milliliter that can successfully infect host cells and express a reporter gene, expressed as TU/mL (transducing units/mL) or IU/mL (infectious units/mL). It directly reflects the vector’s infectivity — insufficient infectious titer leads to low transduction efficiency, compromising therapeutic efficacy.
Common Assays: TCID50, Flow Cytometry, ICA, Live-Cell Analysis
• TCID50 (50% Tissue Culture Infective Dose): Serially dilute AAV samples and infect sensitive cells with the aid of a helper virus. After 48–72 hours, detect reporter gene expression (e.g., GFP) via immunofluorescence. Calculate the dilution at which 50% of cells are infected to determine the infectious titer. Traditional “gold standard”, but subjective (manual counting of fluorescent cells) and time-consuming (3–5 days).
• Flow Cytometry: For AAV carrying a fluorescent reporter gene, infected cells are analyzed by flow cytometry to determine the proportion of positive cells. Combined with dilution factors, infectious titer is calculated. Highly objective (automated counting) and can simultaneously assess cell viability (to exclude cytotoxic effects). Increasingly replacing TCID50 as the mainstream method.
• ICA (Infectious Center Assay): Uses AAV-rep and cap stable cell lines as hosts, without requiring co-infection with wild-type AAV, reducing contamination risk. After infection, “replication centers” form, detectable via hybridization or immunofluorescence. Positive cells are counted under a microscope.
• Live-Cell Analysis: Captures real-time microscopic images of cells before and after transduction to assess infectious titer, providing dynamic insights into gene expression and cellular physiology.
3.2 Target Sequence Expression Level
Expression levels are evaluated at both mRNA and protein levels.
• mRNA Level (qPCR): Measures transcription efficiency. mRNA levels directly reflect the transcriptional activity of the target sequence. The core goal is to confirm successful transcription of the therapeutic gene into functional mRNA and quantify transcript abundance, avoiding transcriptional failure due to promoter inactivity or sequence mutations.
• Protein Level (WB, ELISA, Immunostaining): Evaluates translation efficiency and functional protein expression. Protein-level analysis confirms the presence, quantity, and activity of the therapeutic protein, ensuring that codon usage bias, abnormal post-translational modifications, or other translation issues do not compromise protein function or activity.
III. Purity: Reducing “Ineffective/Harmful Components” and Lowering Immune Risk
The purity of AAV vectors directly impacts clinical safety. Impurities such as aggregated particles or host cell residues can trigger immune responses (e.g., complement activation, antibody production). Empty capsids, while non-toxic, compete for cell surface receptors, reducing the efficiency of functional vector transduction, and higher viral load increases the risk of immune activation.
Therefore, purity control focuses on three key metrics: capsid purity, empty capsid ratio, and aggregation rate, quantified using high-resolution separation techniques.
1. Capsid Purity: Eliminating “Non-Capsid Protein Impurities”
Capsid purity refers to the proportion of capsid proteins relative to total protein, requiring removal of impurities such as host cell proteins (HCPs), bovine serum albumin (BSA, from culture media), and proteases (used during purification). These impurities are primary sources of immunogenicity (e.g., HCPs can induce anti-HCP antibodies). Common methods:SDS-PAGE, ELISA, LC-MS/MS: SDS-PAGE with silver staining:Observes VP1/VP2/VP3 bands and confirms the absence of non-specific bands. Weak bands are further identified by Western Blot and quantified by ELISA. ELISA:Uses antibodies specific for host cell proteins (HCPs from HEK293 or Sf9 cells) or plasmid-encoded proteins (e.g., Rep protein). LC-MS/MS:Currently the most powerful and comprehensive method for detecting and identifying non-capsid proteins, considered the “gold standard” for in-depth characterization.
2. Empty Capsid Ratio: Controlling the Proportion of Non-Functional Particles
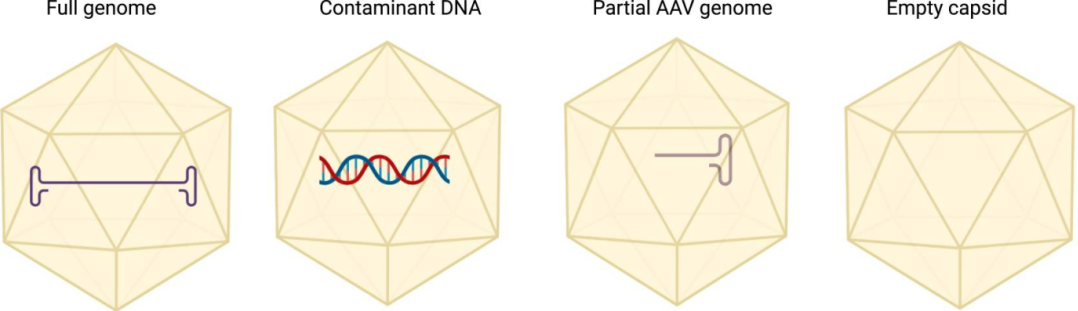
During AAV production, besides full-length, genome-containing vectors, empty capsids, partially filled capsids, overpackaged genomes, host DNA fragments, plasmid DNA fragments, and nucleases may also form.
Empty capsids are an unavoidable byproduct of AAV assembly (capsids without genome or with incomplete genome). Their surfaces are identical to full capsids, competing for cell receptors and potentially activating the immune system (e.g., complement). Therefore, empty capsid ratio is a critical quality control parameter.
Figure 1. Schematic of the various AAV products that can be found in a production batch (PMID: 39234444).
Common methods: TEM (Transmission Electron Microscopy): Directly visualizes capsid populations, providing qualitative and semi-quantitative assessment.
AUC (Analytical Ultracentrifugation):Separates empty, partially filled, and full capsids under centrifugal force, monitored in real time by optical systems (UV or interference), generating sedimentation coefficient distributions to quantify empty vs. full capsids. Current gold standard, but long (24–48 h) and costly; mainly used for clinical or registration batches.
CDMS (Charge Detection Mass Spectrometry): Converts AAV particles into charged ions via electrospray, measuring charge and mass to directly calculate empty/full capsid ratios and distinguish partially filled capsids. Emerging advanced technique included in FDA’s “advanced testing methods” list, potentially the future first-choice method.
IEC/AEX (Ion Exchange Chromatography/Anion Exchange):Separates full and empty capsids based on surface charge differences (full capsids have more negative charge due to encapsidated DNA). Peak areas calculate empty capsid ratio. Fast (1–2 h), low-cost, accuracy ≥85%, suitable for pilot and commercial batch release, but buffer ionic strength must be controlled.
Capsid aggregation is another critical impurity that affects AAV stability, safety, and transduction efficiency. Common methods: DLS, SEC
DLS (Dynamic Light Scattering):Measures particle size distribution in solution, detecting early aggregation before visible precipitation.
SEC (Size-Exclusion Chromatography):Separates and quantifies aggregated versus monomeric AAV particles. Often combined with multi-angle light scattering (SEC-MALS) to accurately determine particle size.
IV. Impurities: Precise Control of “Sources of Risk”
Various impurities can arise during AAV production, requiring targeted detection and control strategies.
1. Host Cell–Derived Impurities
Include host cell DNA (HCD), host cell proteins (HCPs), and other residues.
2. Process-Related Impurities
Include residual nucleases, plasmid DNA, and other process-related components.
3. Common Analytical Methods
ELISA (Enzyme-Linked Immunosorbent Assay): Uses a sandwich format with dual antibodies to detect residual HCPs, e.g., from HEK293 cells. qPCR (Quantitative Real-Time PCR): Detects residual host cell DNA, plasmid DNA, and the size of residual DNA fragments. HPLC (High-Performance Liquid Chromatography): Detects residual lysis reagents, transfection reagents, and other process-related chemicals.
V. Safety: The “Baseline Requirement” Throughout the Product Lifecycle
Safety is a core objective of AAV quality control and must cover the entire production process and the product’s lifecycle. Contaminants such as mycoplasma, endotoxins, and adventitious agents pose significant safety risks and require rigorous testing.
Adventitious agents are unintended viral or microbial contaminants that may arise during cell culture or vector production. Mycoplasma contamination can alter AAV potency and cell viability, while endotoxins (lipopolysaccharides from Gram-negative bacteria) can trigger severe immune responses.
1. Sterility Testing
Uses direct inoculation and membrane filtration methods with appropriate culture media to detect aerobic bacteria, anaerobic bacteria, and fungi. This is mandatory for all injectable biologics.
2. Mycoplasma Detection Methods
PCR: Primers targeting conserved regions of 16S and 23S rRNA are used, followed by agarose gel electrophoresis. Culture-Based Methods: Traditional culture on agar or liquid media is commonly employed. Indicator Cell Culture Assay:Samples are co-cultured with indicator cells (e.g., Vero), stained with DNA dyes (e.g., Hoechst 33258), and examined under a fluorescence microscope for fluorescent particles representing mycoplasma DNA. This method is faster (3–5 days).
3. Bacterial Endotoxin Detection
LAL (Limulus Amebocyte Lysate) Assay: The endotoxin reacts with LAL to produce a coagulation enzyme that cleaves a synthetic chromogenic substrate. The amount of yellow p-nitroaniline formed is measured to determine endotoxin levels. This assay is considered the gold standard for endotoxin quantification.
4. Bacterial Identification
16S rRNA Gene PCR: Molecular identification of bacteria is achieved by PCR amplification and sequence analysis of characteristic 16S ribosomal RNA gene sequences.
5. Viral Contamination Testing
PCR and qPCR: Use specific primers and probes to amplify and detect nucleic acids of known adventitious viruses. Cytopathic Effect (CPE) and Hemadsorption Assays:Samples are inoculated onto multiple indicator cell lines (e.g., MRC-5, Vero). Observe cytopathic effects and perform hemadsorption tests with guinea pig red blood cells to detect viruses capable of red blood cell agglutination (e.g., parainfluenza virus). These are classical pharmacopeial methods. Animal Inoculation:Samples are injected into neonatal mice, adult mice, or chicken embryos, and animals are monitored for disease symptoms or mortality.
Summary
The CQA of AAV vectors form the scientific foundation for their quality control and batch release. From identity verification of the capsid and genome, to multi-level potency assessment including genomic titer and infectious titer, and further to precise control of empty capsid ratio, aggregation, and various process-related impurities, every step directly impacts product efficacy and safety.
With continuous advancements in analytical technologies—such as ddPCR, CDMS, and NGS—AAV quality control is evolving toward higher resolution, automation, and real-time monitoring. Establishing and continuously improving a rigorous, scientifically sound, and regulatory-compliant CQA evaluation system is essential for successfully translating AAV gene therapy products from the laboratory to commercial application.
References
Kontogiannis T, Braybrook J, McElroy C, et al. Characterization of AAV vectors: A review of analytical techniques and critical quality attributes. Mol Ther Methods Clin Dev. 2024;32(3):101309. Published 2024 Jul 30. doi:10.1016/j.omtm.2024.101309
Park MT, Verma A, Froelich CA, Motevalian SP. Process and quality considerations for recombinant adeno-associated virus manufacturing platforms. Trends Biotechnol. 2025;43(8):1921-1937. doi:10.1016/j.tibtech.2025.02.016
Center for Drug Evaluation, National Medical Products Administration (NMPA). Technical Guidelines for Pharmaceutical Research and Evaluation for Clinical Trial Applications of Recombinant Adeno-Associated Virus Vector–Based In Vivo Gene Therapy Products. January 2024.
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Quality Standards: Test Methods and Acceptance Criteria for Biotechnology Products and Biologicals. March 1999.
Service Type :
Select the service you'd like to purchase.
Order Information(Premade-AAVs)
Please provide us some information about the service you'd like to order.
Order Information(Custom AAV/Lentivirus)
Please provide us some information about the service you'd like to order.
Order Information(Others)
Please provide us some information about the service you'd like to order.