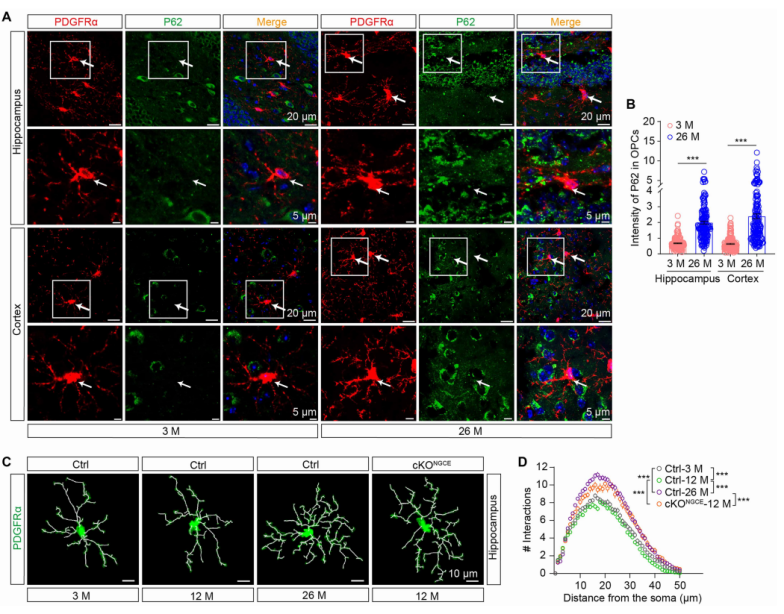
To validate the specific signaling axis OPC autophagy defect → CCL3/CCL5–CCR5 → cognitive decline, the researchers implemented three intervention strategies: Senolytic treatment with dasatinib + quercetin to eliminate senescent cells. Neutralization of SASP factors using anti-CCL3 and anti-CCL5 antibodies. CCR5 inhibition with maraviroc (MVC) to block downstream signaling.
All three interventions reversed electrophysiological abnormalities and rescued cognitive deficits, confirming that OPC autophagy deficiency leads to cognitive decline via the CCL3/CCL5–CCR5 pathway.
3. Application of Patch-Clamp Technology
Patch-clamp recording served as the core experimental technique in this study, providing high-resolution measurements of electrophysiological activity in individual neurons and synapses. This approach allowed researchers to translate the cellular-level autophagy defects in OPCs into quantifiable neuronal functional abnormalities.
3.1 Preparation for Patch-Clamp Experiments
Brain slice preparation:
Mice were deeply anesthetized with isoflurane, and their brains were rapidly removed and placed in ice-cold artificial cerebrospinal fluid (ACSF). Coronal slices (300 μm thick) were prepared using a vibratome. ACSF was continuously bubbled with 95% O₂ and 5% CO₂ to maintain pH 7.4–7.45 and osmolarity 305–315 mosmol. Slices were equilibrated in ACSF at room temperature for at least one hour before recording to allow neuronal recovery. During recording, slices were perfused with oxygenated ACSF to ensure a stable environment.
Electrode setup and recording modes:
Two main patch-clamp modes were used according to the electrophysiological parameters examined:
Voltage-clamp mode: for recording postsynaptic currents (sEPSCs, sIPSCs, mEPSCs). The membrane potential was held constant; pipettes contained CsCl (to block K⁺ currents and reduce interference) with resistance of 2–5 MΩ.
Current-clamp mode: for recording action potentials (sAPs, eAPs). Membrane potential was not fixed; pipettes contained KCl to mimic intracellular ionic conditions, enabling direct recording of membrane potential changes and neuronal firing.
3.2 Core Recording Parameters
To comprehensively assess the effects of OPC autophagy deficiency on neuronal function, five key electrophysiological parameters were evaluated across three dimensions—synaptic transmission efficiency (sEPSCs, sIPSCs, mEPSCs), neuronal excitability (sAPs, eAPs), and synaptic plasticity (LTP).
(1) Glutamatergic and GABAergic Synaptic Transmission
Using voltage-clamp mode, CA1 pyramidal neurons were held at –80 mV (to record excitatory sEPSCs) and +10 mV (to record inhibitory sIPSCs). Spontaneous synaptic events were recorded for 5–10 minutes.
Results showed that, compared with control mice, middle-aged cKONGC mice had a significantly reduced sEPSC frequency but unchanged amplitude (Fig. 3B–D), while sIPSC frequency and amplitude remained unaffected (Fig. 3E–G).
These findings indicate that autophagy deficiency in OPCs selectively impairs glutamatergic transmission without affecting inhibitory synapses.
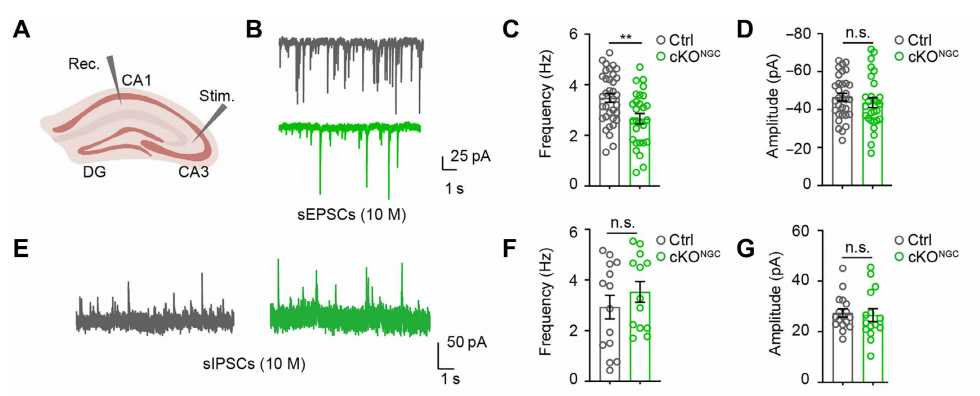 Fig. 3. Effects of OPC autophagy deficiency on hippocampal CA1 synaptic transmission.
Fig. 3. Effects of OPC autophagy deficiency on hippocampal CA1 synaptic transmission.
(2) Miniature Excitatory Postsynaptic Currents (mEPSCs): Presynaptic Mechanism
To isolate presynaptic mechanisms, slices were incubated in ACSF containing 1 μM tetrodotoxin (TTX) for at least 5 minutes to block Na⁺ channels and action potentials. Only random neurotransmitter release events were recorded.
The mEPSC frequency was significantly reduced in cKONGCE mice, while the amplitude was unchanged (Fig. 4N–S), suggesting that impaired glutamatergic transmission results from reduced presynaptic neurotransmitter release probability, not postsynaptic receptor dysfunction.
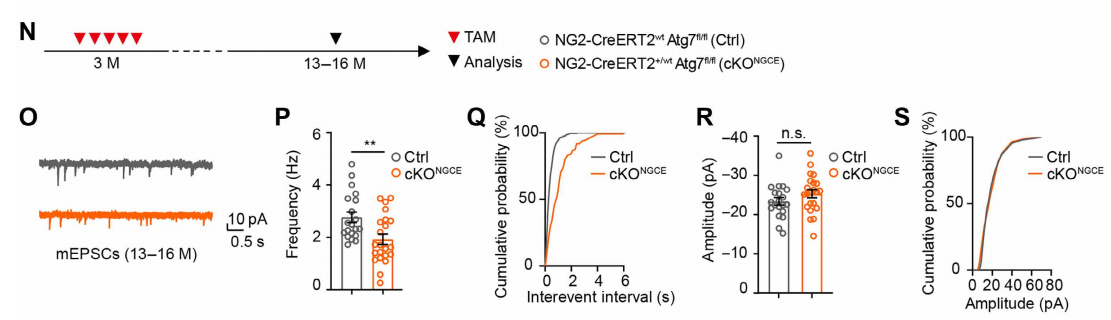
Fig. 4. Effects of OPC autophagy deficiency on presynaptic transmission in hippocampal CA1 neurons.
(3) Spontaneous and Evoked Action Potentials (sAPs, eAPs): Neuronal Excitability
In current-clamp mode, neuronal firing was recorded under two conditions:Spontaneous APs (sAPs): baseline excitability without external stimuli. Evoked APs (eAPs): induced by 200 ms current pulses (20–200 pA, 20 pA steps) to assess stimulus responsiveness.
Results showed that sAP frequency was reduced in cKONGC mice, while amplitude was unchanged (Fig. 5H–J), indicating diminished intrinsic excitability. However, eAP number and amplitude were comparable between groups (Fig. 5K–M), implying normal intrinsic firing machinery and that excitability loss stems from reduced presynaptic input.
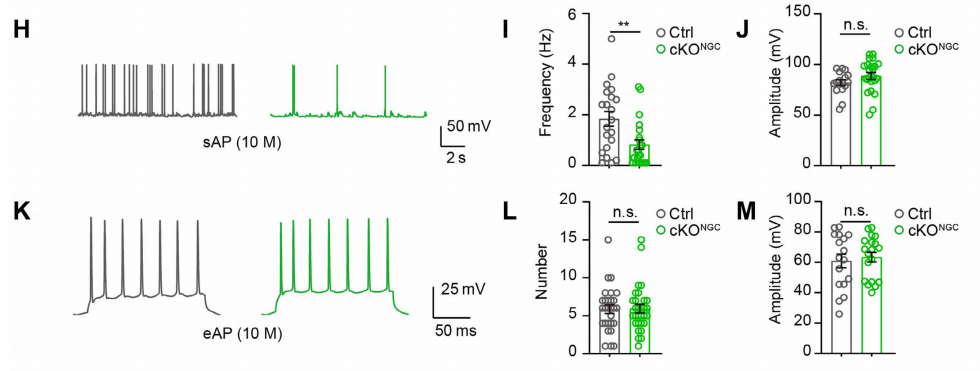 Fig. 5. OPC autophagy deficiency reduces spontaneous but not evoked neuronal firing in hippocampal CA1 neurons.
Fig. 5. OPC autophagy deficiency reduces spontaneous but not evoked neuronal firing in hippocampal CA1 neurons.
(4) Long-Term Potentiation (LTP): The “Cellular Indicator” of Cognitive Function
Stimulation electrodes were placed in the CA3 Schaffer collateral (presynaptic fibers), and recording electrodes in the CA1 stratum radiatum (postsynaptic region). Baseline field EPSPs (fEPSPs) were recorded at 0.033 Hz, and LTP was induced with four trains of 100 Hz stimulation for 1 s each, spaced 20 s apart. fEPSP slope and amplitude were tracked for 60 minutes.
Results showed that cKONGCE mice exhibited reduced fEPSP peak slope and incomplete recovery after 60 minutes (Fig. 6T–V), indicating long-term synaptic plasticity impairment.
Additional analyses: Input–output (I/O) curves assessed overall synaptic strength (0.05–1.0 mA). Paired-pulse ratio (PPR) (150 ms interval) measured presynaptic Ca²⁺ release probability.
PPR was significantly decreased in cKONGCE mice (Fig. 6W–Y), suggesting that OPC autophagy defects reduce presynaptic Ca²⁺ release and thereby suppress LTP.
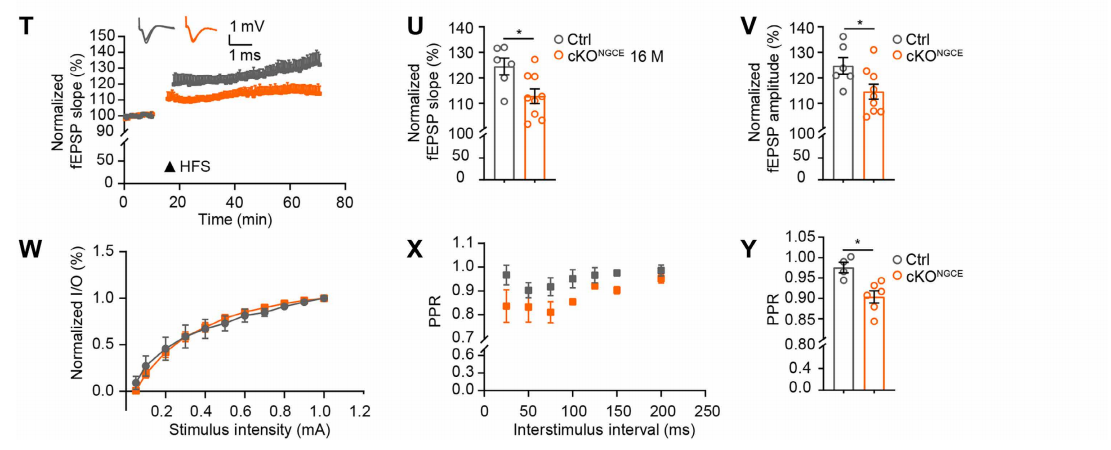 Fig. 6. Impaired LTP in hippocampal CA1 neurons due to OPC autophagy deficiency.
Fig. 6. Impaired LTP in hippocampal CA1 neurons due to OPC autophagy deficiency.
(5) Mechanistic Validation: Blocking CCL3/CCL5–CCR5 Reverses Electrophysiological Deficits
To verify the signaling mechanism, acute brain slices from cKOPαCE mice were incubated with anti-CCL3 and anti-CCL5 antibodies for 20 minutes before recording sEPSCs, sAPs, and LTP. The treatment increased sEPSC frequency (Fig. 7I–K), enhanced sAP frequency (Fig. 5L–N), and restored LTP (Fig. 7O–Q)—fully reversing the electrophysiological abnormalities.
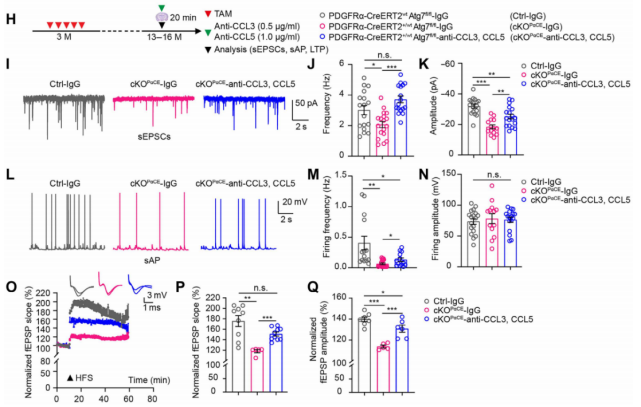 Fig. 7. Anti-CCL3/CCL5 antibodies rescue electrophysiological deficits in hippocampal neurons of cKOPαCE mice.
Fig. 7. Anti-CCL3/CCL5 antibodies rescue electrophysiological deficits in hippocampal neurons of cKOPαCE mice.
Similarly, incubation with the CCR5 antagonist maraviroc (MVC, 15 nM) for 20 minutes reversed deficits in glutamatergic transmission (Fig. 8F–H), neuronal excitability (Fig. 6I–K), and LTP (Fig. 8L–N). These findings confirm that neuronal plasticity suppression caused by autophagy-deficient OPCs is mediated through activation of the CCR5 signaling pathway.
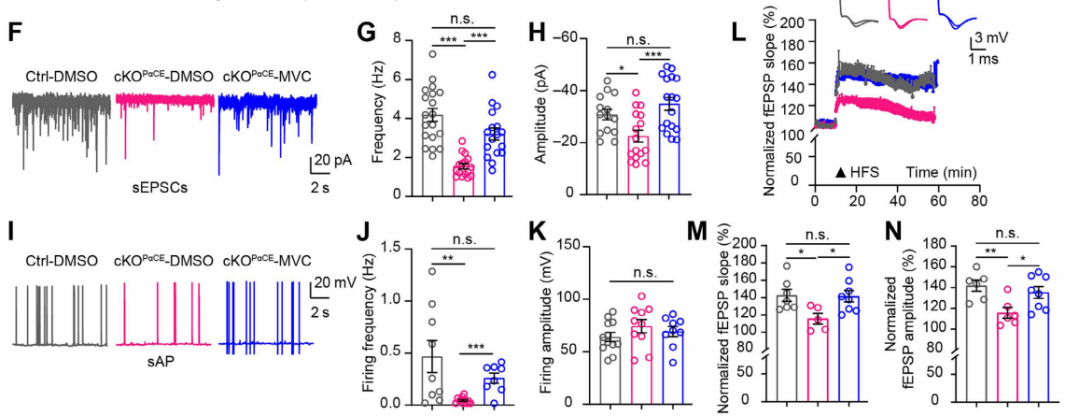 Fig. 8. CCR5 antagonist maraviroc reverses electrophysiological impairments in hippocampal neurons of cKOPαCE mice.
Fig. 8. CCR5 antagonist maraviroc reverses electrophysiological impairments in hippocampal neurons of cKOPαCE mice.
4. Conclusion
By integrating all experimental findings, this study elucidates a complete mechanistic pathway through which autophagy deficiency in OPCs accelerates cognitive decline during brain aging:
Decreased autophagic flux in OPCs of aged mice → Impaired clearance of senescence-related cellular waste, leading to OPC senescence → Senescent OPCs secrete SASP factors CCL3 and CCL5 → CCL3/CCL5 bind to CCR5 receptors on glutamatergic neurons → Inhibition of presynaptic Ca²⁺ release → Impaired glutamatergic transmission, reduced neuronal excitability, and suppressed LTP → Decreased neuronal plasticity → Ultimately resulting in cognitive decline
Importantly, this entire process is independent of the myelination function of OPCs (Fig. 9).
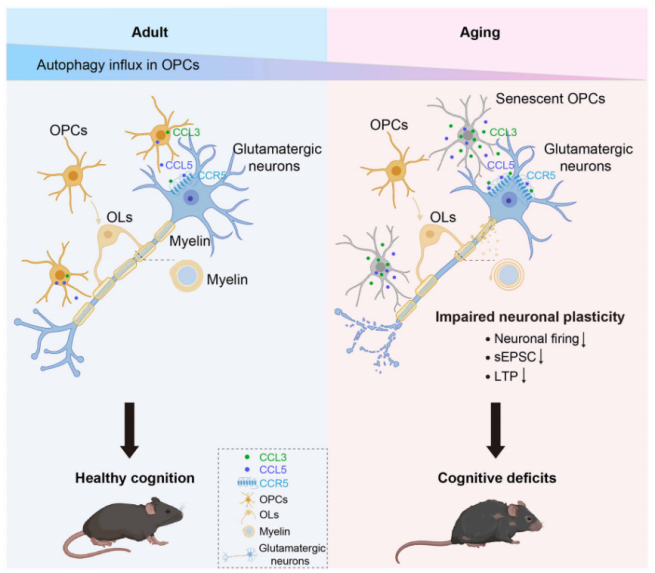
Fig. 9. Schematic illustration of how OPC autophagy deficiency accelerates brain aging and cognitive decline.
Contact Us
Brain Case Biotech provides comprehensive electrophysiological testing services aligned with the experimental approaches described in this study.
Our services include brain slice preparation, experimental protocol optimization, and quantitative data analysis, ensuring precise solutions tailored to various neuroscience research needs. Our technical team will provide dedicated support — bd@ebraincase.com
References
Chen H, Sun YY, Li QF, et al. Impaired macroautophagy in oligodendrocyte precursor cells suppresses neuronal plasticity via a senescence-associated signaling. Sci Adv. 2025 Sep 26;11(39):eadq7665. doi: 10.1126/sciadv.adq7665. Epub 2025 Sep 24. PMID: 40991686; PMCID: PMC12459396.