- E-mail:BD@ebraincase.com
- Tel:+8618971215294
To study the function of a gene, experiments are often designed from two directions: gain of function and loss of function. Currently, CRISPR and RNA interference (RNAi) technologies are the primary methods used to explore the loss of gene function. Unlike CRISPR gene knockout, RNAi technology does not alter the genome sequence of the target cells but instead regulates gene expression at the transcriptional level by specifically targeting one or more mRNA transcripts of the target gene. Therefore, this technique is widely applied in gene function research, as well as in the treatment of cancer and viral infectious diseases.
In gene knockdown experiments, two main methods are commonly used: chemically synthesized small interfering RNA (siRNA) and vector-based short hairpin RNA (shRNA). Both types of RNA molecules can specifically bind to the mRNA of the target gene and guide its degradation, thereby reducing the expression level of the target gene.
siRNA is a double-stranded RNA molecule. Once introduced into cells, siRNA forms an RNA-induced silencing complex (RISC) with specific enzymes in the cytoplasm. Within RISC, siRNA is unwound into single strands, with the antisense strand specifically recognizing and binding to the target gene’s mRNA. The mRNA is then cleaved, leading to its degradation and reduced protein translation, thus regulating the expression of the target gene. On the other hand, shRNA is an RNA molecule that forms a hairpin structure. Inside the cell, shRNA is processed into siRNA, which then performs RNA interference (RNAi) functions.
Next, let’s walk through the specific experimental design and operational workflow.
Determine the target gene that needs to be silenced based on the research objective. A gene may have multiple abbreviations, but for consistency, each gene in the NCBI database has a unique Gene ID. Transcript information and the CDS sequence of the target gene can be found on the NCBI website (link: https://www.ncbi.nlm.nih.gov/gene).
Based on the sequence of the target gene, design and synthesize siRNA or shRNA that specifically binds to the target gene. The former involves directly synthesizing a siRNA sequence (19-27 bp) targeting the gene, which is then transfected into cells for expression. The latter involves converting the siRNA into shRNA (encoding both the sense and antisense strands of the siRNA, separated by a loop sequence), and constructing an expression vector for shRNA, using plasmids or viruses to express it in cells or in vivo.
Before designing the RNA molecule, choose between siRNA or shRNA depending on the experimental goal. The main differences between the two are as follows:

Table 1. The main differences between siRNA and shRNA
The design of shRNA can be facilitated using online tools, such as those provided by Sigma, Life Technologies, and GPP. The Sigma website offers a comprehensive shRNA library for humans and mice, and some shRNA sequences for specific genes have been validated for direct use, making the process convenient and efficient. For example, to target the human TP53 gene:
a. Visit the website: https://www.sigmaaldrich.com/US/en/semi-configurators/sirna?activeLink=selectAssays. Enter the gene name or Gene ID and click "Search".

Fig 1. The "Predesigned siRNA" tool website-1
b. Select Species - Human or Others - Confirm. Review the sequence information (some shRNA sequences in the website have been validated, and you can choose those with verified high knockdown efficiency).
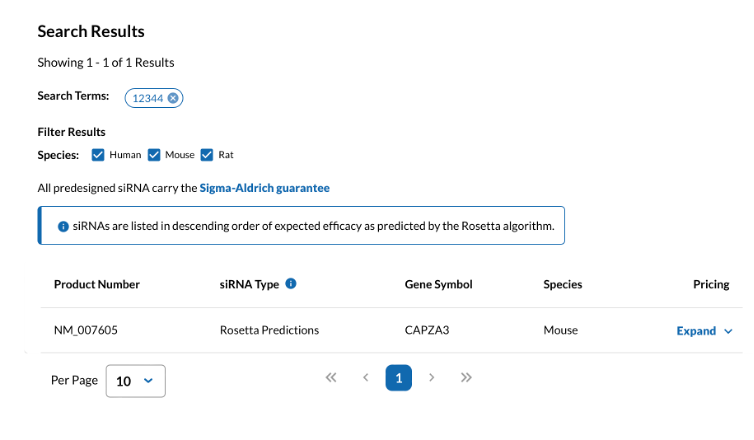
Fig 2. The "Predesigned siRNA" tool website-2
c. Perform a BLAST search of the target sequence from the website against the target gene on the NCBI website (link: https://blast.ncbi.nlm.nih.gov/Blast.cgi). Ensure that the designed shRNA sequence specifically targets the target gene and has more than 3 mismatched bases with other genes.
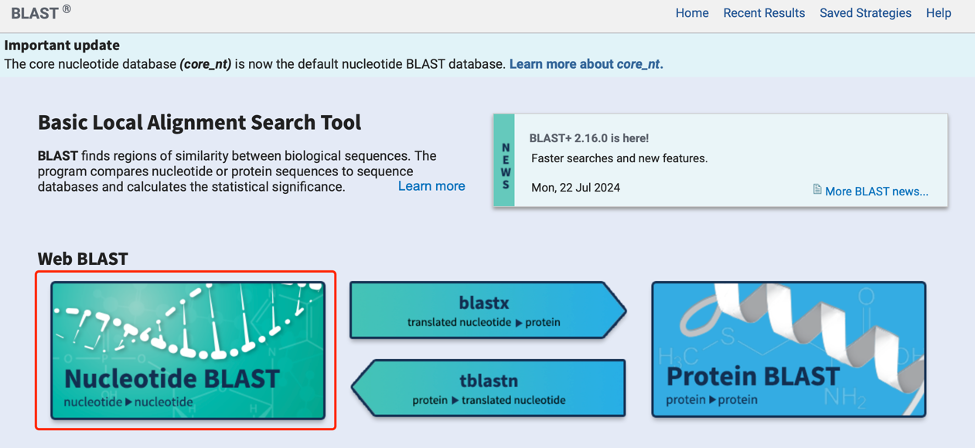
Fig 3. NCBI-BLAST
If gene silencing is achieved through direct transfection of siRNA, chemical synthesis is sufficient. However, if siRNA needs to be delivered for long-term expression or in vivo using plasmids or viruses such as lentivirus (LV) or adeno-associated virus (AAV), it is necessary to construct an appropriate core plasmid in advance and produce the corresponding plasmid or viral particles for expression in cells or in vivo. Choosing the right expression vector is crucial. For example, with AAV:
shRNA is a type of small/short hairpin double-stranded RNA. Due to the short construction sequence, a specific pol III promoter like U6 or H1 is used for in vivo expression. The U6/H1 promoter is known for its strong expression capability and broad range, making it suitable for gene interference in the entire brain or tissue. For example, the vector structure for an AAV core plasmid is as follows:

Figure 4. Example Structure of AAV-U6-shRNA Expression Vector
In many research studies, specific knockdown in a certain tissue or cell is needed. Since U6 and H1 do not have tissue specificity, a tissue-specific promoter is required for gene silencing in specific tissues. Researchers use the miRNA and shRNA's similar synthetic mechanisms, using pre-miRNA as a backbone to produce siRNA, allowing for longer shRNA sequences within the miRNA backbone. This enables the use of pol II promoters (including various tissue-specific promoters like hSyn, GFAP) for specific tissue gene silencing [1]. Common miRNA backbones for gene interference include miR30, miR155, and miR17-92, with miR30 being the most frequently used. For example, the vector structure for an AAV core plasmid is as follows:
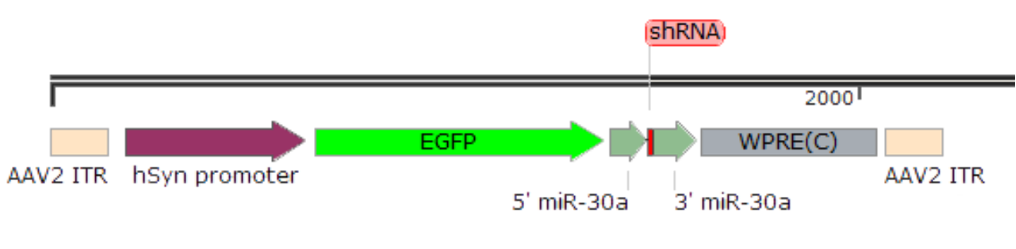
Figure 5. Example Structure of AAV-miR30-shRNA Expression Vector
-Specificity Through Promoters: Allows for specific expression in different tissues or cells based on the characteristics of various pol II promoters. This includes not only broad-spectrum promoters like CMV, CAG, EF1α but also tissue-specific promoters like hSyn.
-Co-expression of Multiple shRNAs: Vectors based on the miR30 backbone can express two shRNAs simultaneously (targeting one gene with two sites or two different genes), with minimal difference in expression levels between the two shRNAs.
-Retention of Natural miR30 Circular Structure: Ensures precise Dicer cleavage and reduces off-target effects.
-Cre and Flp Dependent Expression: miR30 backbone vectors can incorporate recombinase loxP sites, allowing use with Cre viruses or Cre mice for expression that is specific and regulated by Cre expression.
Introduce the designed shRNA or siRNA into cells via transfection or other methods. Within the cells, shRNA will be transcribed and expressed, leading to the formation of siRNA. After allowing sufficient time for expression, extract the RNA and protein to assess the knockdown effect of the target gene. Techniques such as real-time quantitative PCR (qRT-PCR), Western Blot, and immunohistochemistry can be used to measure the expression levels of the target gene at both RNA and protein levels, ensuring successful suppression of the target gene expression.
Brain Case Biotech not only provides custom interference viruses for conventional genes but also offers custom interference viruses for non-coding RNAs such as circRNA, IncRNA, and microRNA. Custom viruses can include shRNA design, shRNA vector construction, and validation of shRNA interference efficiency in vitro. If you have any design issues or packaging requirements for such AAVs, please contact BD@ebraincase.com
Q: The target gene found in NCBI has 3 mRNAs, but siRNA can only be designed for one mRNA. The existing literature does not provide guidance on this. How should I choose?
A: If the 3 mRNAs produce at least 3 different proteins, select the mRNA corresponding to the protein of interest. If the mRNAs do not produce proteins, choose the mRNA based on its independent functional relevance. If the goal is to study the gene's overall function, design siRNAs targeting each mRNA to study them separately.
Q: Negative controls should have no homology with the target gene's sequence. How should transfection efficiency be assessed?
A: Negative controls can include a fluorescence protein tag to indicate transfection. Cells should exhibit fluorescence if transfected successfully, which helps optimize transfection conditions and evaluate efficiency. Interference efficiency is related to shRNA design.
Q: For cellular level experiments, should I choose a vector-expressed shRNA or small fragment siRNA? What are the differences in transfection efficiency and silencing duration?
A: Choose between shRNA and siRNA based on the duration of silencing needed. For short-term inhibition (up to 1 week), siRNA is preferable as it does not require vector construction, saving time, and can be used with multiple transfections to extend RNAi effects up to 2 weeks. For long-term knockdown (over 2 weeks), shRNA expression vectors are better due to prolonged expression, though they require vector construction. Efficiency primarily depends on shRNA design and varies with cell type and experimental conditions.
Q: I'm starting an experiment to stably express RNAi fragments in cells. Which is better and more efficient: shRNA or siRNA?
A: For stable transfection, shRNA is the preferred choice. If the transfection efficiency of the cells is low, plasmids can be packaged into lentivirus to improve infection efficiency.
Q: Will placing two shRNAs targeting different genes in the same vector interfere with each other?
A: When using a vector to express two shRNAs, the key issue is whether both shRNAs are expressed and processed successfully. This largely depends on the vector and its expression efficiency. Protein interactions between the targets depend on whether the two proteins affect each other.
Q: Can any target gene be used for stable cell line creation with lentivirus-mediated RNA interference? How long does the screening process typically take?
A: Not all genes are suitable for stable cell line creation. Two prerequisites must be met: the cells must be able to stably survive and proliferate, and the target gene's mRNA must be effectively silenced. If silencing the mRNA impairs cell survival or stability, a stable cell line cannot be established. The screening process duration varies based on these factors.
Q: For comparing cellular behaviors before and after RNA interference of a specific gene, is siRNA or shRNA more efficient?
A: In terms of interference efficiency alone, there is no significant difference between siRNA and shRNA. However, for transfection efficiency, shRNA, delivered via plasmids or viruses, can ensure that 100% of cells are knocked down through selection. SiRNA, often used with transfection reagents, may introduce cytotoxicity that affects results. If the target protein is critical, this could lead to significant differences. siRNA is costlier and usually used for emergency situations or as a complement. ShRNA is generally preferred for its cost-effectiveness and ability to provide stable models, although it may have limitations such as potential phenotype attenuation over time.

WhatsApp Business Account
Address:-
Address:-



