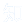听力损失是老年人高发疾病,已被公认为认知衰退的最重要可干预危险因素之一,但二者的因果关系尚不明确。成年海马神经发生对认知功能至关重要,且对环境因素敏感,但听力损失是否通过影响海马神经发生导致认知障碍尚不清楚。
2026年3月19日,中国科学院遗传与发育生物学研究所郭伟翔团队在Cell Stem Cell期刊上(IF=20.4)在线发表题为“Auditory activity sustains adult neurogenesis and cognition through the locus coeruleus-norepinephrine system”的研究论文。该研究首次揭示,听力损失通过削弱脑桥尾侧网状核谷氨酸能神经元(PnCᵛᴳˡᵘᵀ²)→蓝斑去甲肾上腺素能神经元(LCNEergic)的神经通路,降低海马齿状回(DG)去甲肾上腺素水平,损害成年神经发生,进而导致认知衰退,为听力损失与痴呆的因果关系提供了直接证据。

研究利用Slc26a5DTR/+小鼠通过白喉毒素(DT)特异性消融耳蜗外毛细胞(OHCs),成功构建不伴随应激与中枢炎症的重度听力损失模型,c-fos免疫组化表明选择性消融外毛细胞可特异性损伤听觉环路的神经元活动。为明确听力损失与认知衰退的因果关系,对DT处理后的Slc26a5DTR/+小鼠进行系列行为学检测(图1A)。结果显示,与对照组相比,DT处理组小鼠在Y迷宫(图1B、1C)、新物体位置测试(图1D、1E)、新物体识别测试(图1F,G)及情境恐惧条件反射测试(图1H、1I)中表现均显著下降,表明其短期记忆、空间记忆、物体识别记忆及联想学习能力受损;而在旷场实验(图1J-L)、高架十字迷宫(图1M-O)及悬尾实验(图1P、1Q)中两组无显著差异。综上,听力损失仅特异性导致认知功能障碍,不影响运动能力、焦虑样行为及抑郁样行为。
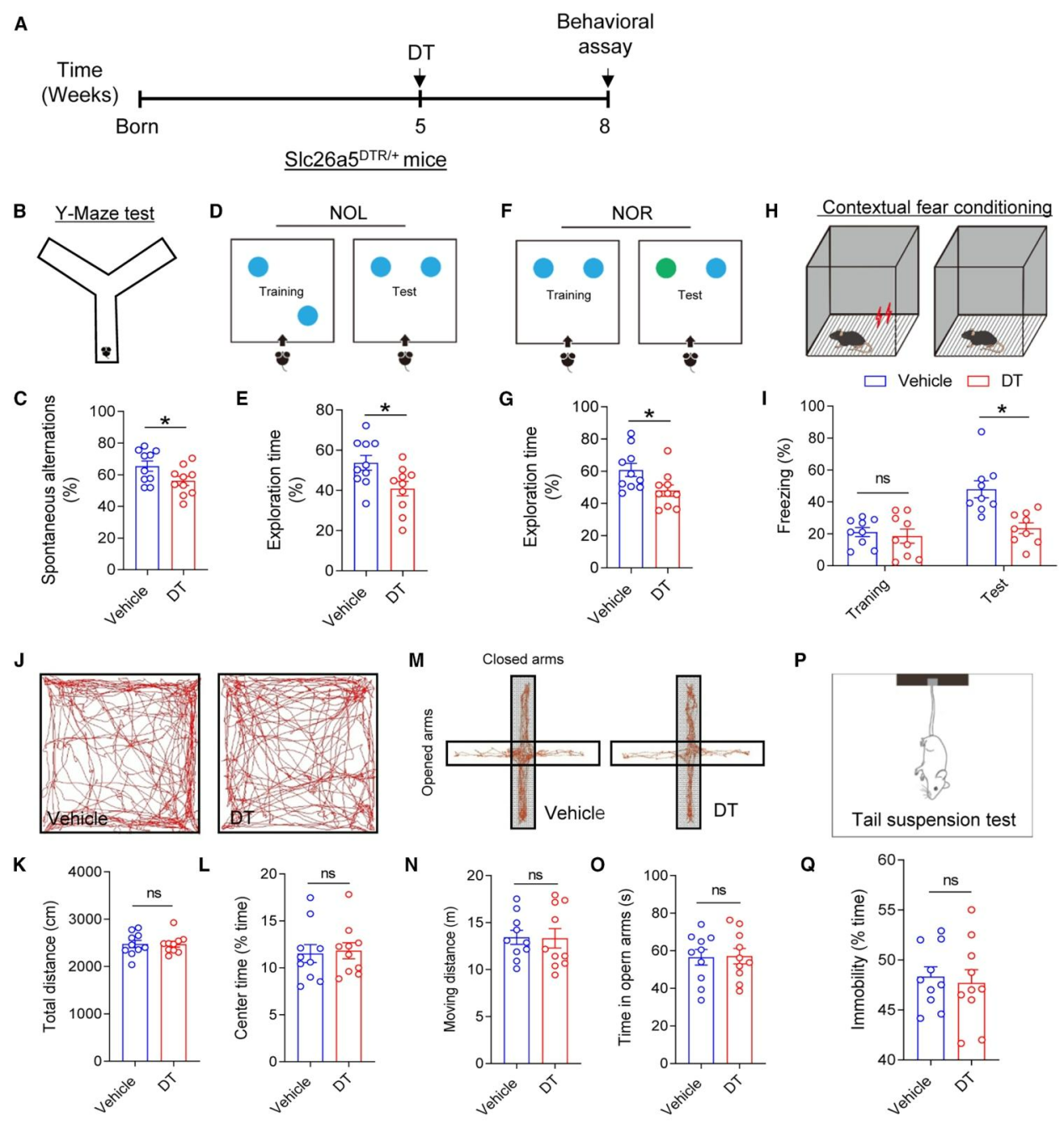
图1.特异性敲除耳蜗OHCs基因可导致认知功能障碍,但不引起运动能力异常或焦虑样行为
为探究听力损失对成年海马神经发生的影响,研究者将Slc26a5DTR/+小鼠与Nestin-GFP小鼠杂交获得Slc26a5DTR/+::Nestin-GFP小鼠,并于5周龄注射DT,3周后检测发现,DT处理组海马齿状回(DG)中Nestin-GFP阳性细胞及放射状胶质样神经干细胞(RGLs)数量显著减少(图2A-D),Mcm2(增殖标志物)标记的增殖细胞与Mcm2+Nestin-GFP+双阳性增殖型神经干细胞亦明显降低(图2E、2F),且caspase-3检测显示细胞凋亡水平无明显变化(图2G);采用EdU脉冲标记实验评估新生神经元数量(图2H),结果表明DT处理组EdU+NeuN⁺新生神经元数量显著少于对照组(图2I、2J)。综上,听力损失通过抑制神经干细胞增殖,进而减少成年海马神经发生。
图2.特异性敲除耳蜗OHCs会损害成年海马神经发生
已知LC至DG的投射来源于去甲肾上腺素能神经元。为鉴定听觉环路至LCNEergic神经元的突触投射,研究采用逆行单突触示踪技术,在DBH-Cre小鼠LC区域定向注射AAV-DIO-TVA-GFP与AAV-DIO-RG特异性标记去甲肾上腺素能神经元,3周后注射RV-mCherry并于1周后检测;结果显示病毒注射位点精准定位于LC(图3D),LC的传入突触主要来源于脑桥尾侧网状核(PnC)核团(图3E–L),RV标记的PnC神经元胞体直径约20μm,不属于巨型神经元(图3M),且原位杂交证实其主要表达vGluT2而非GlyT2(图3N、3O)。结合逆行AAV示踪及顺向跨单突触HSV示踪技术表明,PnCvGluT2神经元向LC发送兴奋性投射,其突触后靶细胞主要为LC中DBH阳性的去甲肾上腺素能神经元,而非GABA能神经元。综上,LCNEergic神经元直接接收PnCvGluT2神经元的听觉输入。
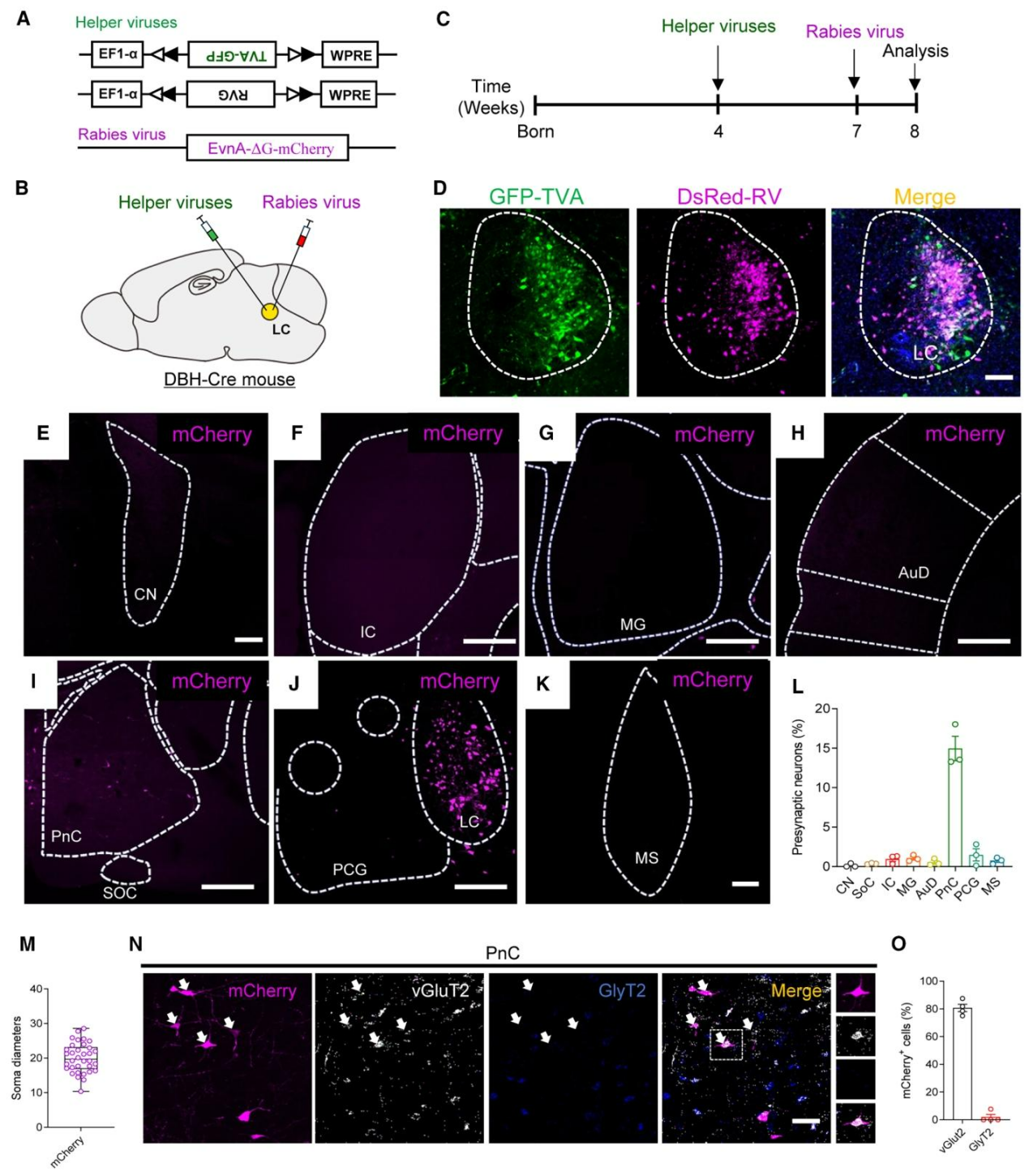
图3.PnCᵛᴳˡᵘᵀ²向LCᴺᴱ能神经元传入的逆行单突触示踪
为明确PnCvGluT2神经元与LCNEergic神经元之间的功能性联系,采用逆行AAV结合化学遗传学与光纤光度记录策略:向DBH-Cre小鼠LC注射retroAAV2-hSyn-Flp及Cre依赖的钙指示剂GCaMP6f,向PnC注射Flp依赖化学遗传学病毒hM3Dq/hM4Di-mCherry(图4A)。结果显示,激活PnCvGluT2神经元可显著升高LCNEergic神经元的GCaMP6f荧光信号,抑制该类神经元则降低其荧光强度(图4B、4C)。进一步在vGluT2-Cre小鼠LC注射retroAAV2-EF1α-DIO-Flp,同时在PnC注射Flp依赖性化学遗传学病毒hM3Dq/hM4Di-mCherry,海马DG注射去甲肾上腺素探针AAV-hSyn-GRAB-NE2h(图4D),光纤光度记录结果显示,激活或抑制PnCvGluT2神经元可相应升高或降低海马DG区的去甲肾上腺素信号,对照组无明显变化(图4E、4F);进一步在DBH-Cre小鼠DG区注射逆行Cre依赖光遗传病毒(NpHR或hChR2),在LC区注射retroAAV2-hSyn-Flp,在PnC区注射Flp依赖性化学遗传学病毒,并在DG区表达去甲肾上腺素探针,以实现对该通路的多层级操控与实时监测(图4G、4K),光抑制该投射可阻断CNO诱导的去甲肾上腺素上升(图4H-J),光激活该投射则能逆转其下降(图4L-N),证明PnCvGluT2→LCNEergic通路可直接调控DG区去甲肾上腺素释放。
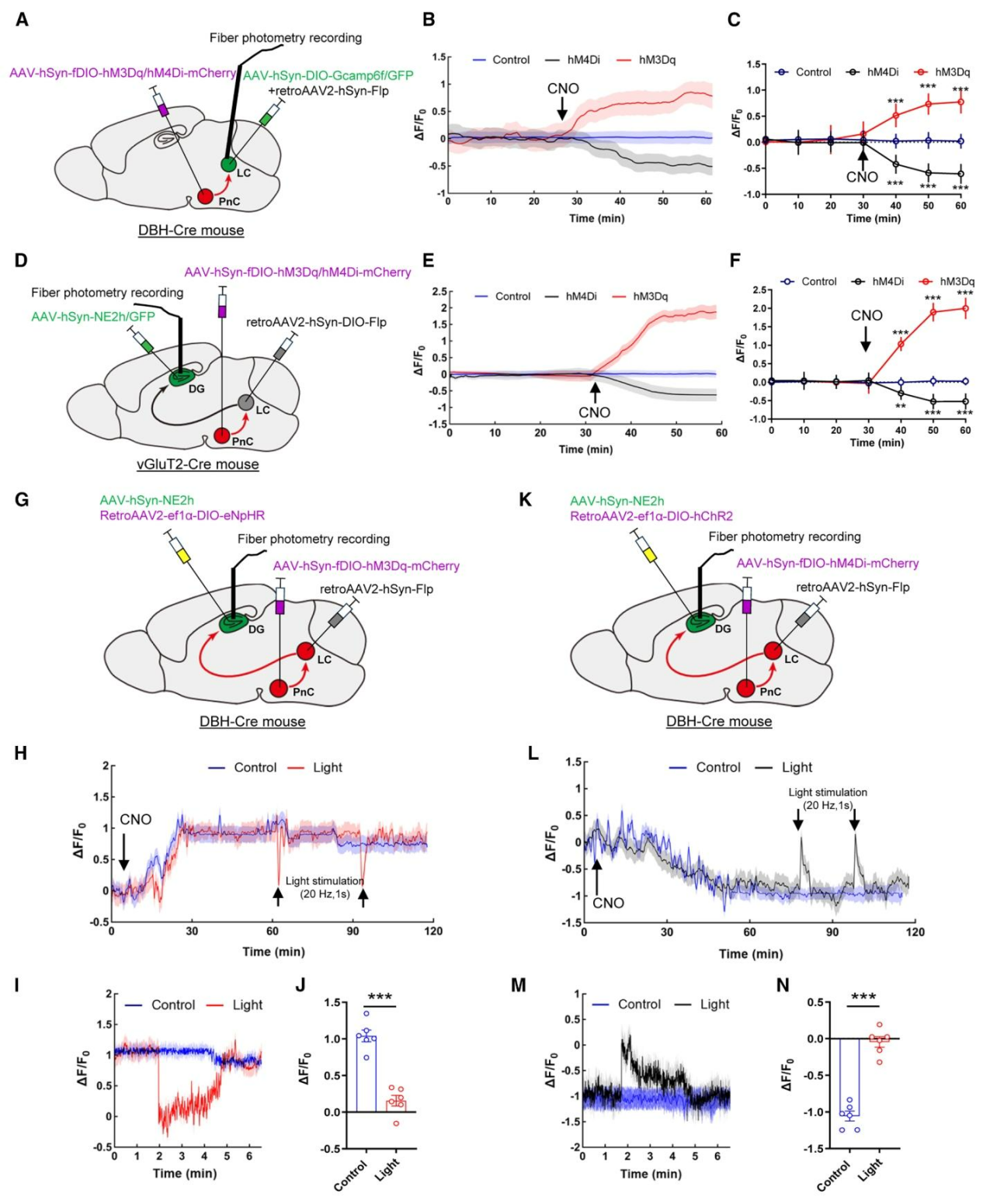
图4. PnCvGluT2→LCNE神经传入调控海马DG的去甲肾上腺素释放
为评估听力损失对神经元活性的影响,研究检测了DT诱导Slc26a5ᴰᵀᴿ/⁺小鼠听力损失后脑内c-Fos的表达情况,发现PnC中vGluT2⁺阳性神经元活性显著降低,而GlyT2⁺阳性神经元无明显变化(图5A-E);同时LC核团内DBH⁺去甲肾上腺素能神经元活性显著下降,GABA⁺神经元活性则无显著差异(图5F-I),这表明听力损失会同时降低PnCvGluT2神经元与LCNE神经元的活性。进一步将AAV-EF1α-DIO-GCaMP6f病毒注射到Slc26a5ᴰᵀᴿ/⁺::DBH-Cre双转基因小鼠LC中(图5J),结果发现,未处理小鼠LCNE神经元可被白噪声刺激显著激活,而听力损失小鼠的LCNE神经元对声音刺激无响应(图5K、5L);利用GRABne2h探针记录海马DG区去甲肾上腺素水平发现,声音刺激可使未处理小鼠DG区去甲肾上腺素信号显著增强,而听力损失小鼠则无明显变化,表明听力损失会降低PnCvGluT2与LCNEergic神经元活性,并进而减少海马齿状回的去甲肾上腺素水平。
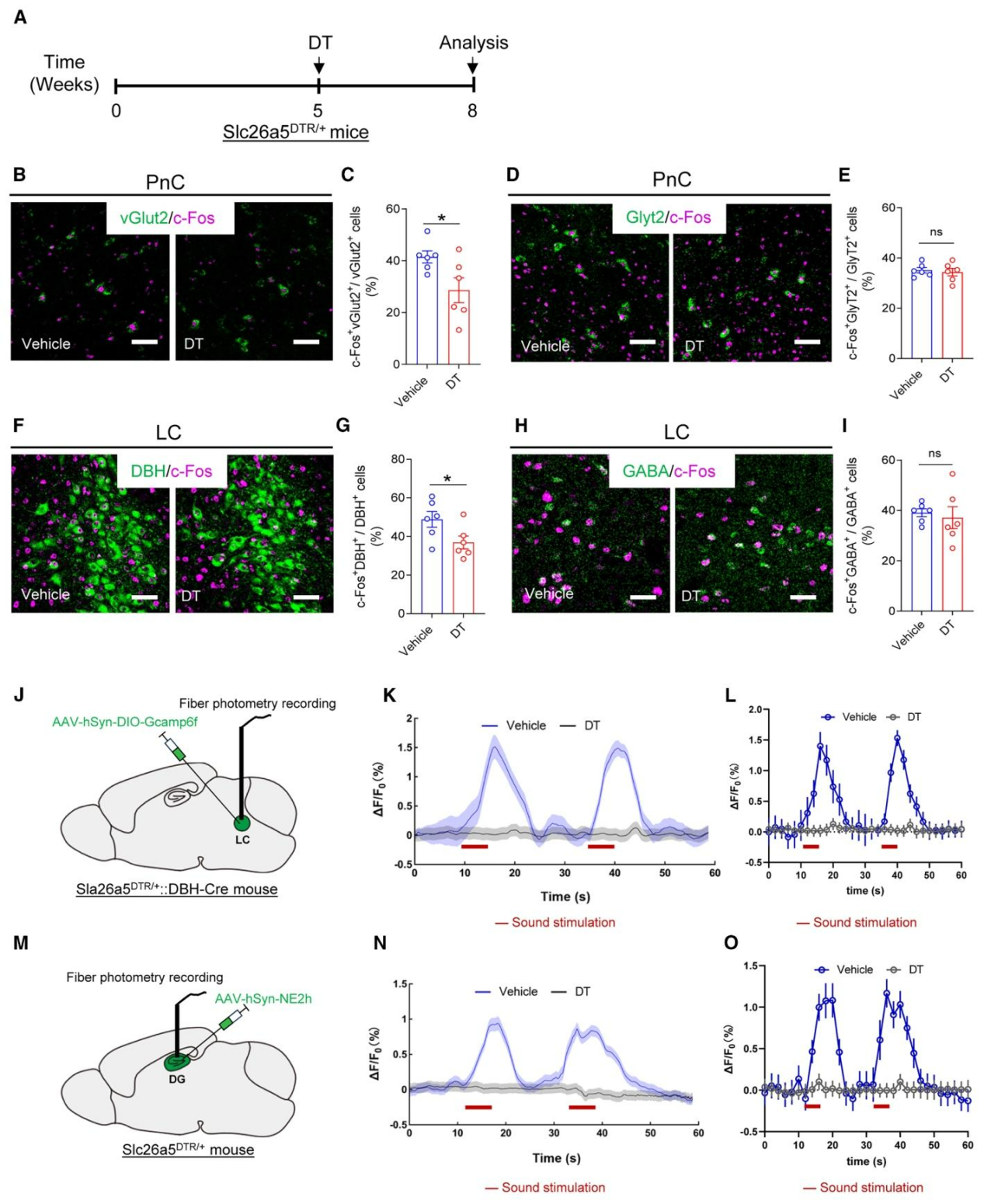
图5. 特异性敲除OHCs会减弱PnCvGluT2向LCNE神经元的神经传入
为探究激活PnCvGluT2→LCNE神经传入是否能够挽救听力损失造成的成年海马神经发生缺陷,研究在DT诱导听力损失的vGluT2-Cre::Slc26a5ᴰᵀᴿ/⁺::Nestin-GFP小鼠LC中注射retroAVV2-EF1α-DIO-Flp、向PnC注射AAV-fDIO-hM3Dq-mCherry,以特异性激活PnCvGluT2→LCNE传入(图6A、6B),CNO处理后,Nestin-GFP⁺细胞数与Nestin-GFP⁺放射状胶质样干细胞(RGLs) 数无明显改变(图6C、6D),但Mcm2⁺增殖细胞与双阳性增殖型RGLs显著增加,(图6E、6F),这提示激活该通路可逆转听力损失所致的神经干细胞增殖抑制;EdU脉冲标记实验表明,CNO处理使经DT处理的vGluT2-Cre::Slc26a5ᴰᵀᴿ/⁺小鼠,小鼠中EdU⁺NeuN⁺成年新生神经元数量显著增加(图6G、6H)。由于神经干细胞可分化为中间祖细胞进而形成新生神经元,推测长期CNO处理会使神经干细胞与祖细胞数量上升。综上,激活PnCvGluT2→LCNE神经传入可挽救听力损失所致的成年海马神经发生缺陷。
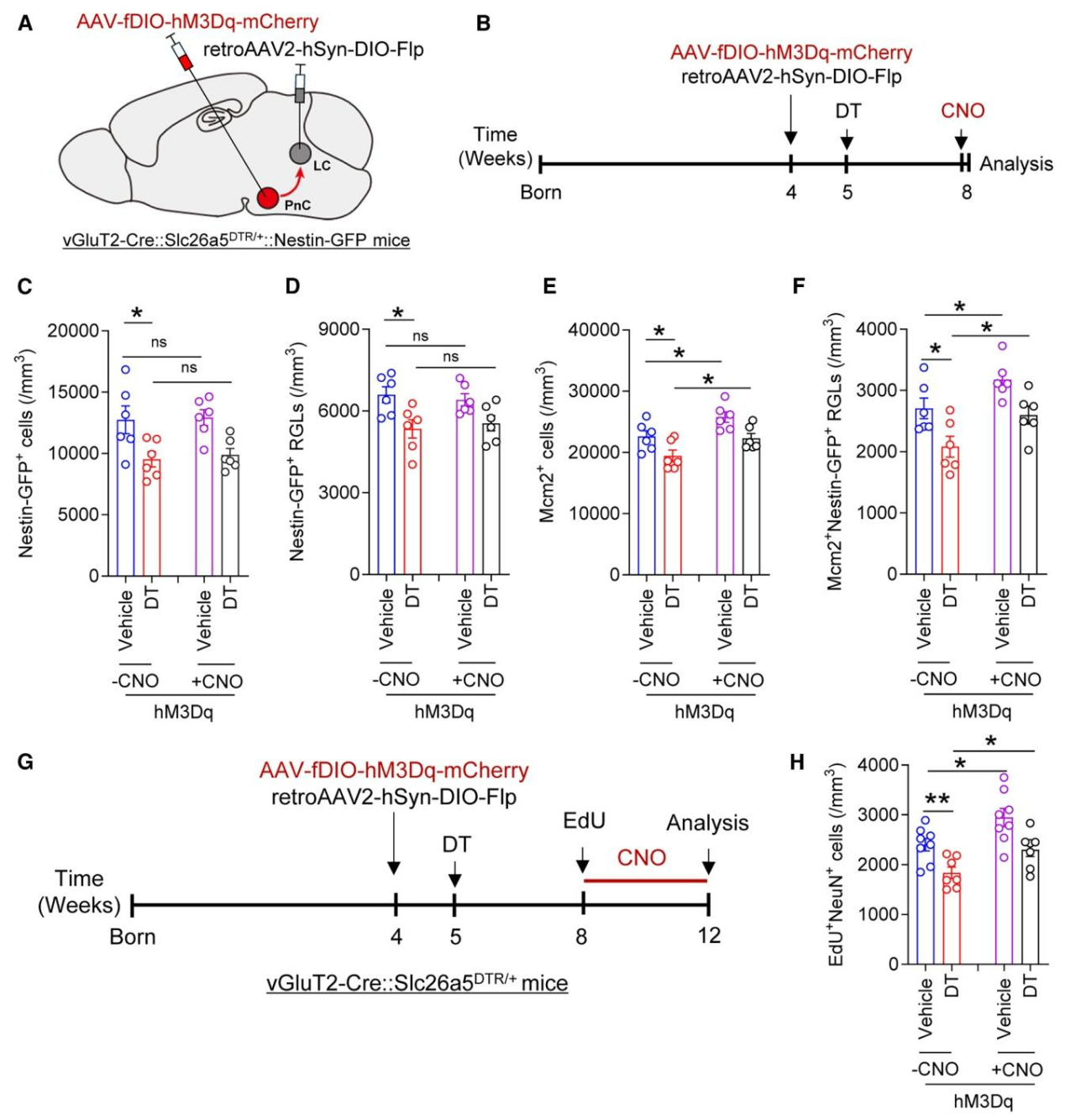
图6. 激活PnCvGluT2→LCNE神经传入可挽救听力损失小鼠的成年海马神经发生缺陷
为探究激活PnCvGluT2→LCNE神经传入是否能够改善听力损失引发的认知功能障碍,研究向DT处理的vGluT2-Cre::Slc26a5ᴰᵀᴿ/⁺小鼠LC注射逆行AAV2-EF1α-DIO-Flp、PnC注射AAV-fDIO-hM3Dq-mCherry,经4周CNO处理后开展行为学检测(图7A、7B),结果显示激活PnCvGluT2→LCNE神经传入可显著改善小鼠的认知功能,表现为Y迷宫自发交替率升高、新物体位置与新物体识别偏好提升、情境恐惧条件反射僵直行为增加(图7C-7F);而海马注射神经发生抑制剂AraC(一种已知可抑制神经发生的抗有丝分裂剂)可完全阻断上述认知改善效应(图7C-7F);但旷场、高架十字迷宫及悬尾实验结果均无明显变化(图7G-7I),这一结果排除了运动能力异常与焦虑样行为在上述过程中的参与,证实激活PnCvGluT2→LCNE传入可通过恢复成年海马神经发生,挽救听力损失导致的认知障碍。
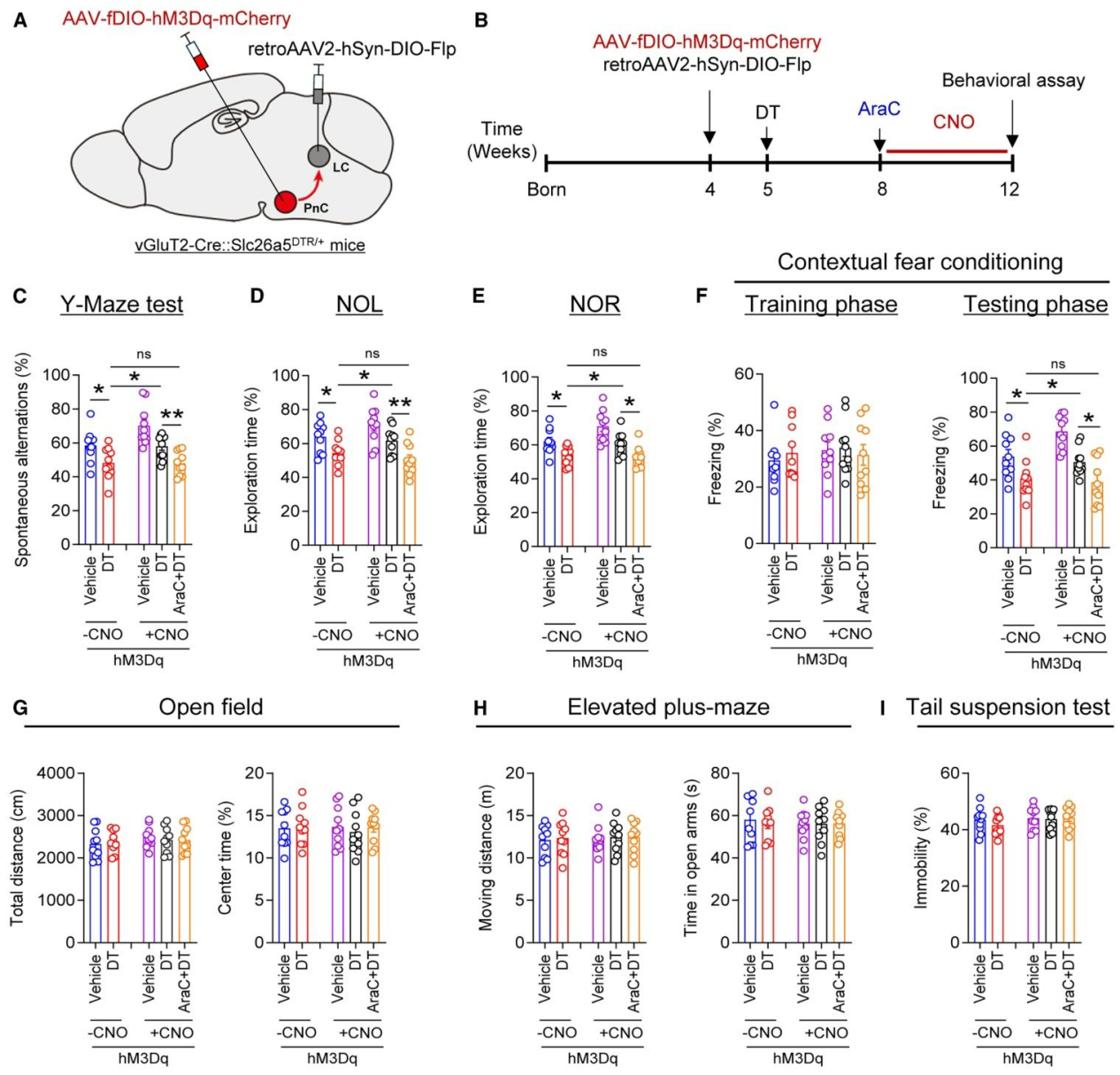
图7. 激活PnCvGluT2→LCNE神经传入可改善听力损失小鼠的认知障碍
本研究首次在神经环路水平明确了听力损失与认知衰退的因果关系:通过特异性敲除耳蜗外毛细胞构建听力损失小鼠模型,发现其认知功能与成年海马神经发生均出现显著缺陷;机制上,听力损失削弱了PnCvGluT2→LCNE神经传入,导致海马去甲肾上腺素水平下降,而激活该通路可挽救上述缺陷,且该改善作用依赖于海马神经发生过程。这一发现揭示了听力损失导致认知障碍的关键神经机制,为相关干预提供了新靶点。
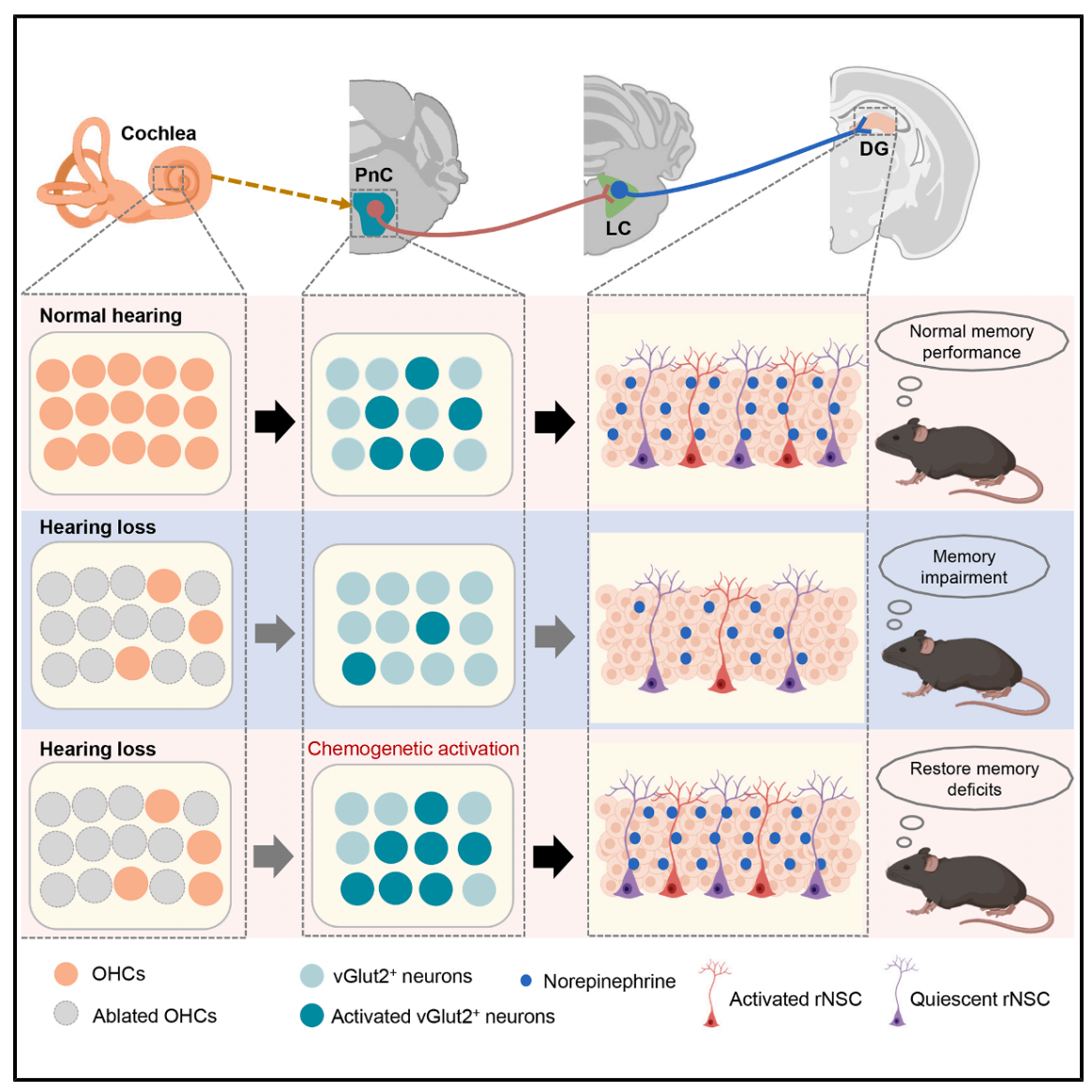
图8. 总结图
本文使用的工具病毒布林凯斯均可提供:
同时布林凯斯也可提供各类定制服务请联系小布:18971216876(微信同号)或者咨询所在区域的销售经理获取更多信息。