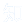胶质母细胞瘤(GBM)是最常见、致死性最高的原发性脑癌,其肿瘤内异质性和浸润性生长模式是导致治疗耐药、术后高复发的核心原因,患者预后极差;GBM细胞可形成肿瘤微管(TM):一种神经突样膜突起,介导胶质瘤细胞间形成互连网络,同时作为神经元-胶质瘤恶性突触的形成位点,接收神经元电化学输入,整合入神经回路并引发脑过度兴奋,但恶性突触形成是否以牺牲正常突触为代价、介导TM形成与突触重排的分泌型信使尚未明确;突触修剪是神经发育/记忆形成的正常过程,研究假设GBM可能劫持该程序,重塑神经微环境以促进自身进展。
2026年2月27日,四川大学华西医院生物治疗研究中心/生物治疗全国重点实验室/国家老年疾病临床医学研究中心汪源/张燕团队,联合清华大学马伟伟/钟毅、江西科技师范大学王松华团队,在肿瘤学国际顶级期刊Cancer Discovery在线发表题为“Glioblastoma-secreted C1QL1 orchestrates tumor microtube expansion and neural synaptic pruning to drive malignant synapse formation and recurrence”相关文章,首次发现GBM分泌的C1QL1是介导胶质瘤-神经元、胶质瘤-胶质瘤交互作用的关键信使,其通过结合受体BAI3激活Rac1信号通路,一方面促进TM扩增、胶质瘤网络形成,另一方面诱导正常神经突触修剪并推动恶性突触形成,最终驱动GBM浸润性生长与复发;研究团队开发的首款非GEF靶向Rac1抑制剂JK50561可穿透血脑屏障,有效阻断C1QL1-BAI3-Rac1轴,同时抑制肿瘤微管形成、挽救突触修剪并减少恶性突触,在GBM术后复发模型中显著延长小鼠生存期,且与放疗联用呈现协同效果。

为了筛选神经元连接性增强的浸润性GBM细胞特征基因,整合多套人类GBM单细胞/空间转录组数据集(GSE117891、GSE223065等)(图1A)。筛选出210个在瘤周区和高功能神经连接区GBM细胞中均高表达的基因(图1B),含少突胶质细胞前体细胞样(OPC样)和神经前体细胞样(NPC样)细胞特征基因,与既往OPC样、NPC样细胞在浸润区富集并形成恶性突触的结论一致(图1A)。其中6个分泌型蛋白基因参与突触及肿瘤微管(TM)/细胞骨架调控,C1QL1为其中唯一与突触修剪相关的基因(图1B),是介导浸润区胶质瘤细胞间及胶质瘤-神经元相互作用的潜在候选分子。C1QL1优先表达于OPC样GBM细胞,不表达于肿瘤相关小胶质细胞/巨噬细胞。多数据集证实,C1QL1在瘤周区、电活性/放电型GBM细胞中高表达(图1C);空间转录组显示,其在肿瘤浸润区、前缘的表达高于实质区(图1D),证实C1QL1在高电活性浸润性GBM细胞中高表达。电活性浸润性GBM细胞与肿瘤复发、不良预后相关,复发性GBM中C1QL1高表达位点及表达水平均高于原发性GBM(图1E)。中国脑胶质瘤基因组图谱(CGGA)和胶质瘤纵向分析研究(GLASS)队列显示,C1QL1表达与复发性GBM患者总生存期负相关(图1F、G),且高表达与复发间隔缩短、复发后生存期降低相关(图1H),证实其预后价值和临床意义。
图1. 多数据集分析确定C1QL1是电活性浸润GBM细胞的标志物
利用患者来源胶质瘤干细胞(GSC)异种移植模型(BNI23、BNI21),研究C1QL1在GBM发生发展中的作用。BNI23 GSC中C1QL1敲除(KO)、BNI21 GSC中C1QL1敲低(KD)两种模型中,C1QL1 KO/KD均能抑制肿瘤发展、延长小鼠生存期(图2A)。通过人核抗原(HNA)和神经丝蛋白(NF200)染色,检测胶质瘤浸润程度以及神经元-胶质瘤邻域的分布情况,显示两种模型终末期肿瘤均浸润周围皮质、沿胼胝体(CC)白质束侵袭,再现人类GBM病理特征,且浸润区(IZ)神经纤维与胶质瘤细胞交织(图2B)。C1QL1 KO/KD可显著降低肿瘤浸润和增殖(图2B、C),而C1QL1过表达则缩短小鼠生存期。为了验证C1QL1致癌作用的遗传保守性,采用TP53/NF1/PTEN三突变人神经干细胞(hNSC)异种移植模型(TNP)和Trp53/Nf1双突变小鼠细胞系(BT5)模型。两种模型中,标记的胶质瘤细胞浸润性强,终末期常侵袭至对侧大脑(图2D)。相反,C1QL1 KO的TNP肿瘤和C1QL1 KD的BT5肿瘤体积缩小、浸润减轻(图2D、E),小鼠生存期延长(图2F);且C1QL1 KO的TNP肿瘤中,OLIG2和HNA双标记的少突胶质细胞谱系肿瘤细胞比例降低,与C1QL1在OPC样GBM细胞中高表达一致。综上所述,不同遗传背景和荷瘤小鼠免疫状态下,C1QL1在多种GBM模型中均能促进肿瘤生长和浸润。
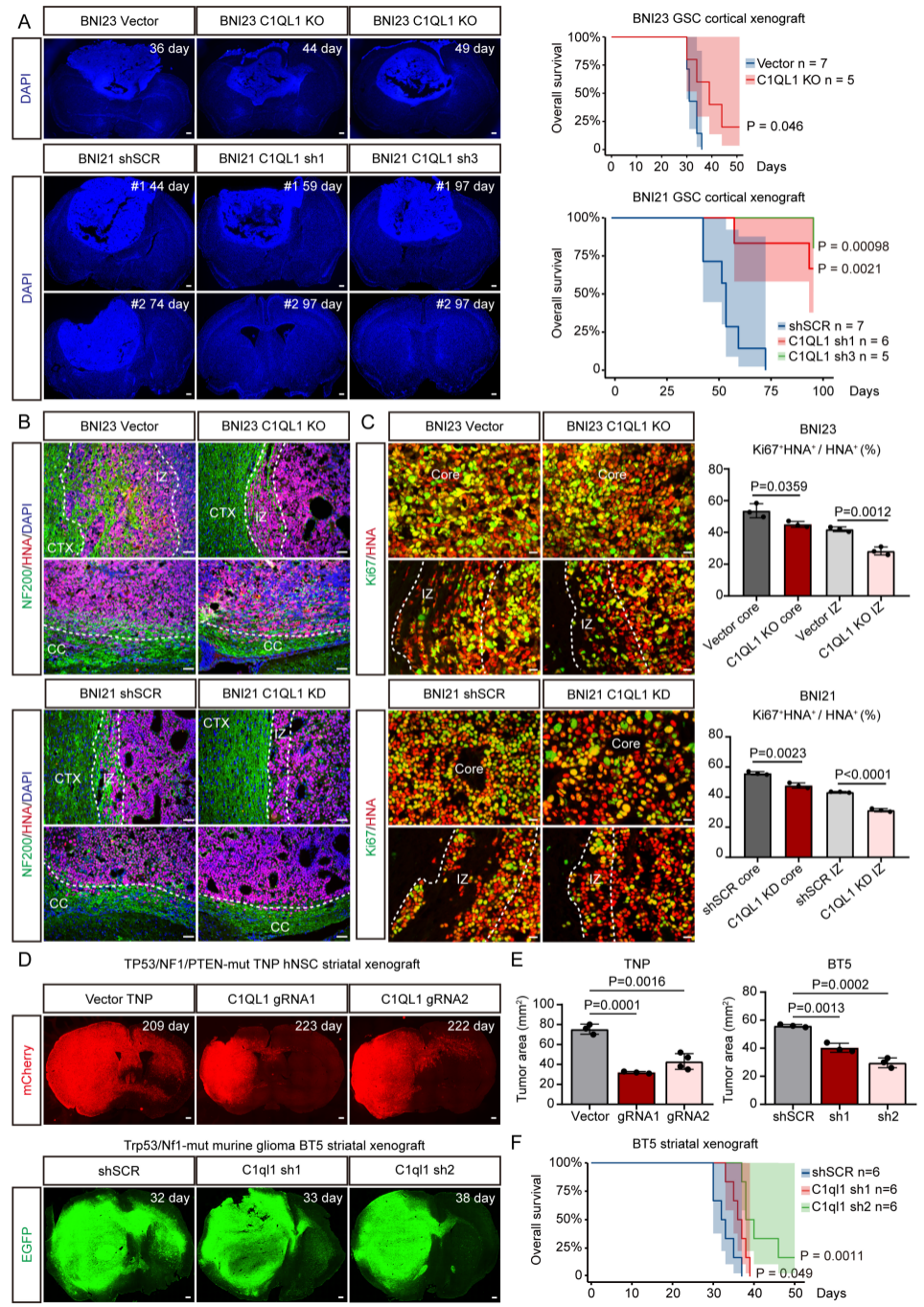
图2. C1QL1在多种GBM模型中促进肿瘤增殖与浸润
鉴于C1QL1在调控神经元突触中的作用,通过单独培养或与原代皮质神经元共培养mCherry+GSC,探究其促瘤效应是否依赖神经元微环境。单独培养时,C1QL1敲除(KO)组GSC每视野平均细胞数较空载体(Vector)组减少37%(图3A);与神经元共培养后,空载体组GSC数量增加36%,C1QL1敲除组无明显变化(图3A)。极限稀释分析(ELDA)显示,C1QL1敲除/敲低(KO/KD)可降低BNI21、BNI23细胞的自我更新及增殖能力(图3B),表明C1QL1对GSC的作用兼具肿瘤细胞内在依赖性和神经元微环境依赖性。为了探究C1QL1的肿瘤细胞内在效应,对空载体组和C1QL1敲除组GSC进行批量RNA测序(bulk RNA-seq)。结果显示,已知肿瘤微管(TM)调控因子在敲除组显著下调(图3C),下调最显著的通路包括细胞连接、跨突触信号传导等(图3D),提示C1QL1可能调控GSC中间隙连接偶联的TM。采用TM标志物人巢蛋白(hNestin)染色验证,两株GSC中,C1QL1敲除组有TM或分支状TM的细胞比例及TM长度均显著降低(图3E);GSC异种移植模型中,敲除组有TM的细胞比例及hNestin标记的TM长度也一致减少(图3F)。因此,C1QL1在体外和体内均能促进TM形成。
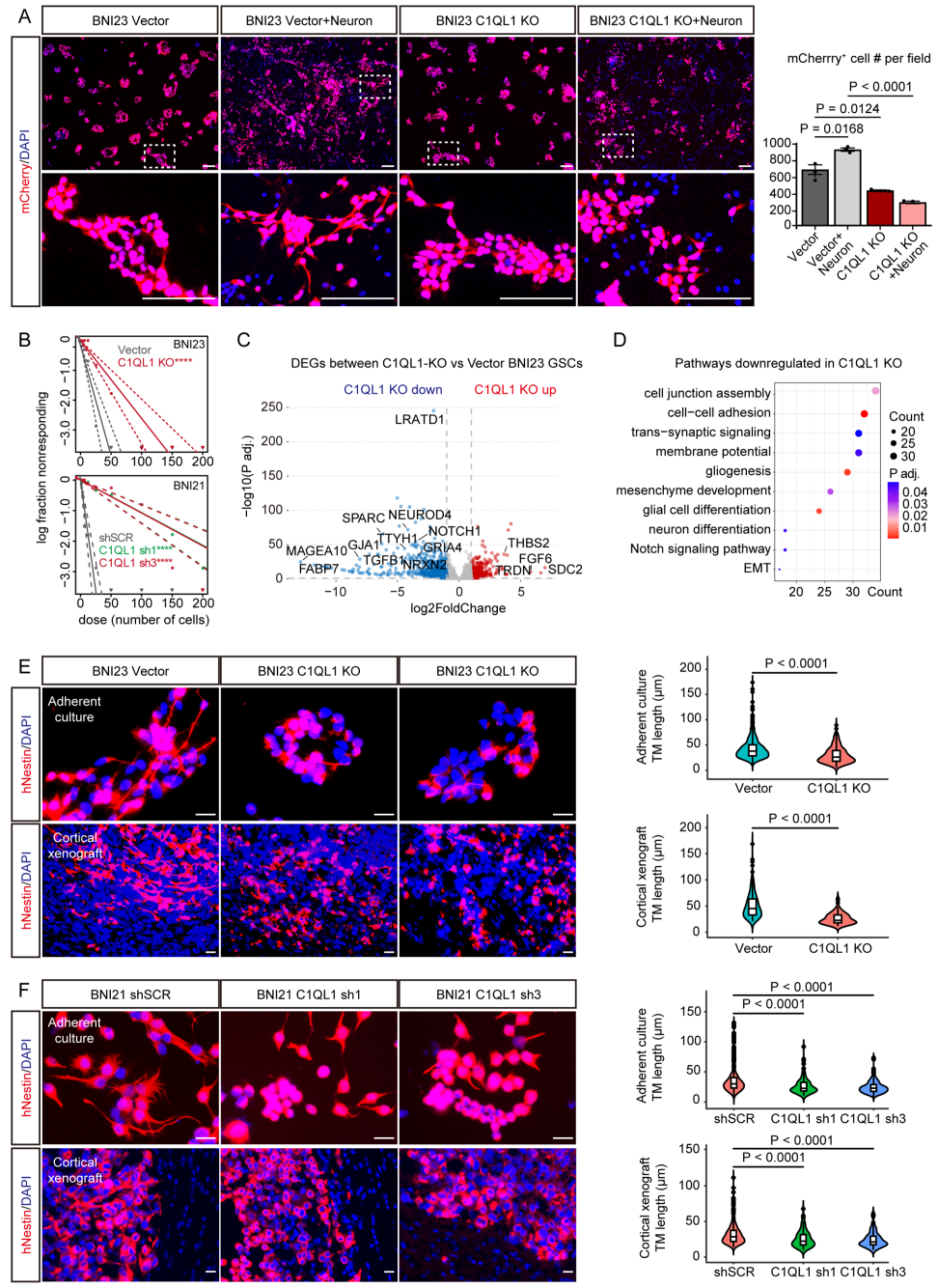
图3. C1QL1促进肿瘤微管形成
对与空载体或C1QL1敲除(KO)GSC共培养的神经元进行批量RNA测序,探究胶质瘤来源C1QL1对神经元的影响。结果显示,C1QL1可促进微环境神经元活性/可塑性;这些下调基因富集于突触可塑性通路上调相关功能(图4A、B)。结合既往C1QL1突触修剪功能,探究其对神经元突触的影响:C1QL1敲除GSC的条件培养基(CM)可提高神经元突触素(Synapsin)阳性突触小点密度(图4C),而C1QL1过表达(OE)GSC的CM则降低突触密度,且该效应可通过稀释CM逆转。钙成像显示,空载体组神经元自发钙瞬变幅度和频率低于C1QL1敲除组(图4D、E),证实C1QL1可诱导神经元突触修剪,减少神经元间电信号传递。
将表达突触后标志物PSD95-GFP的GSC与神经元共培养,定量显示C1QL1敲除组的突触素/PSD95-GFP共定位恶性突触比例,仅为空载体组的约50%(图4F)。mCherry+ GSC皮质异种移植后2~3周,免疫电镜显示两种模型中约11%的胶质瘤突起存在神经元-胶质瘤突触(图4G),C1QL1敲除后恶性突触比例降低约50%(图4H),证实GSC在体内可与神经元形成突触。全细胞膜片钳分析显示,GSC海马异种移植模型中,胶质瘤细胞可记录到刺激诱发的兴奋性突触后电流(EPSC),C1QL1 KO/KD使EPSC幅度显著降低(图4I)。综上,C1QL1可诱导神经元突触修剪,同时促进功能性恶性突触形成。
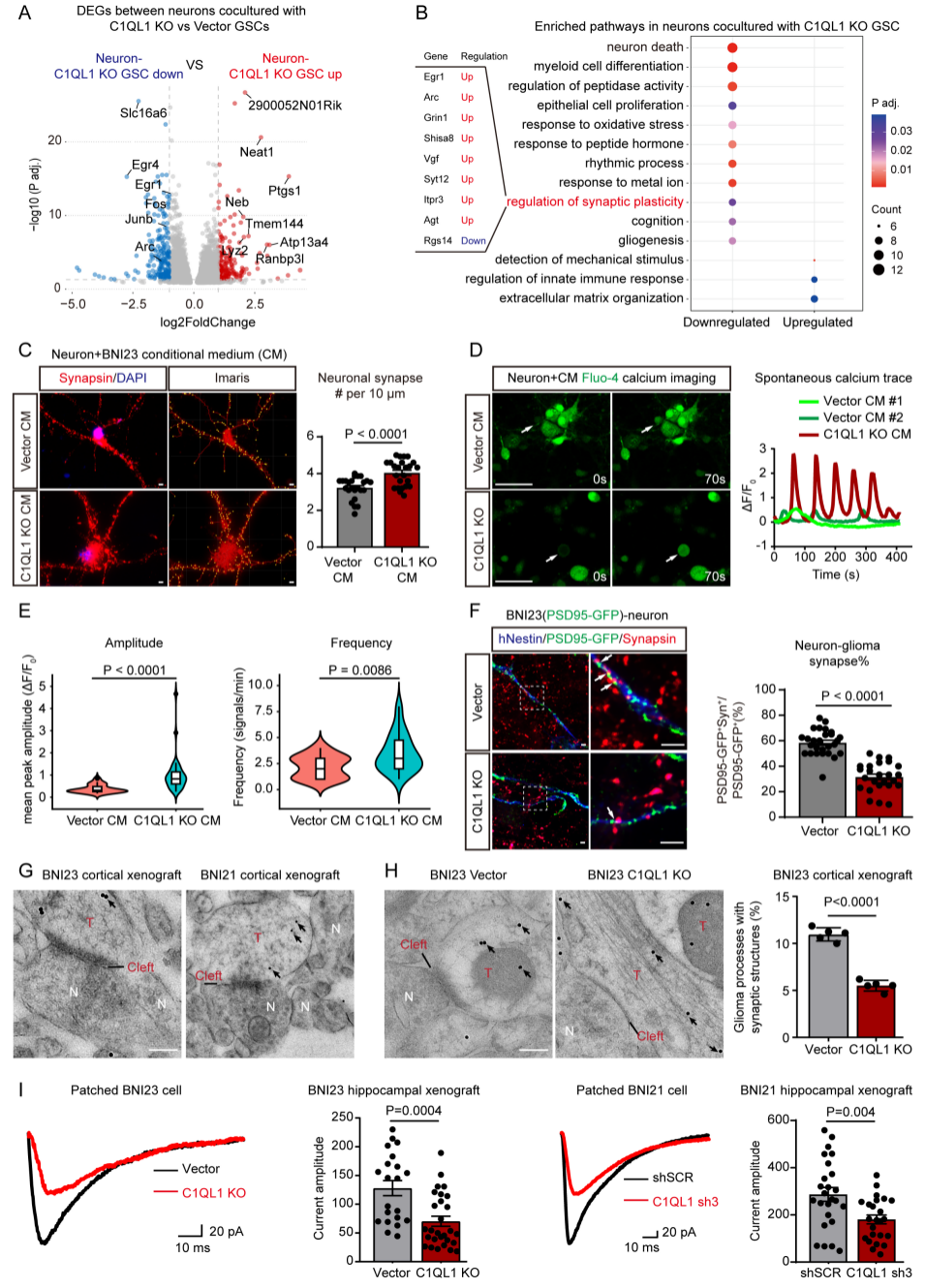
图4. C1QL1在促进恶性突触形成的同时诱导神经元突触修剪
探究了C1QL1的肿瘤细胞内在效应和神经元依赖性效应的分子机制。Flag标记的C1QL1可与其受体BAI3共免疫沉淀,且BAI3在GSC中表达(图5A)。BAI3可通过激活Rho GTP酶Rac1调控树突形态发生,而Rac1是细胞骨架重排和突触可塑性的关键通路。检测显示,两株GSC中C1QL1敲除/敲低(KO/KD)可使Rac1活性(Rac1-GTP比例)降低40%-50%(图5B)。敲低两株GSC中的BAI3,可显著降低Rac1活性(图5C),且其对GSC自我更新/增殖的抑制效应与C1QL1 KO/KD一致,双敲除/敲低无额外抑制作用。此外,BAI3敲低可减少培养GSC及异种移植瘤中TM长度(图5D),与C1QL1 KO/KD效果一致(图3E、F)。因此,BAI3敲低的BNI23 GSC移植小鼠生存期延长(图5E),BNI23 GSC移植后几乎不形成肿瘤。
GSC海马异种移植模型电生理记录显示,胶质瘤细胞BAI3敲低组中,经HNA标记确认的胶质瘤细胞和瘤周神经元的EPSC幅度减弱(图5F),提示恶性突触减少、瘤周神经元过度活性降低。证实C1QL1在胶质瘤细胞中通过BAI3激活Rac1,促进TM和恶性突触形成,推动胶质瘤进展。BAI3在神经元中高表达,GSC分泌的C1QL1-Flag可与共培养神经元树突上的BAI3阳性小点共定位(图5G)。C1QL1过表达(OE)GSC的条件培养基(CM)处理组神经元Rac1活性最高,而C1QL1 KO/KD GSC的CM可降低神经元Rac1活性(图5H)。通过立体定向注射AAV,在小鼠海马/皮质诱导神经元特异性敲低Bai3,再移植野生型GSC。电生理显示,神经元Bai3敲低可降低经HNA标记的胶质瘤细胞EPSC幅度(图5I),且两株GSC模型中,神经元Bai3敲低均能延长小鼠生存期(图5J)。综上,C1QL1-BAI3介导的神经元突触修剪可在体内促进恶性突触形成和肿瘤发展,且独立于其肿瘤细胞内在效应。
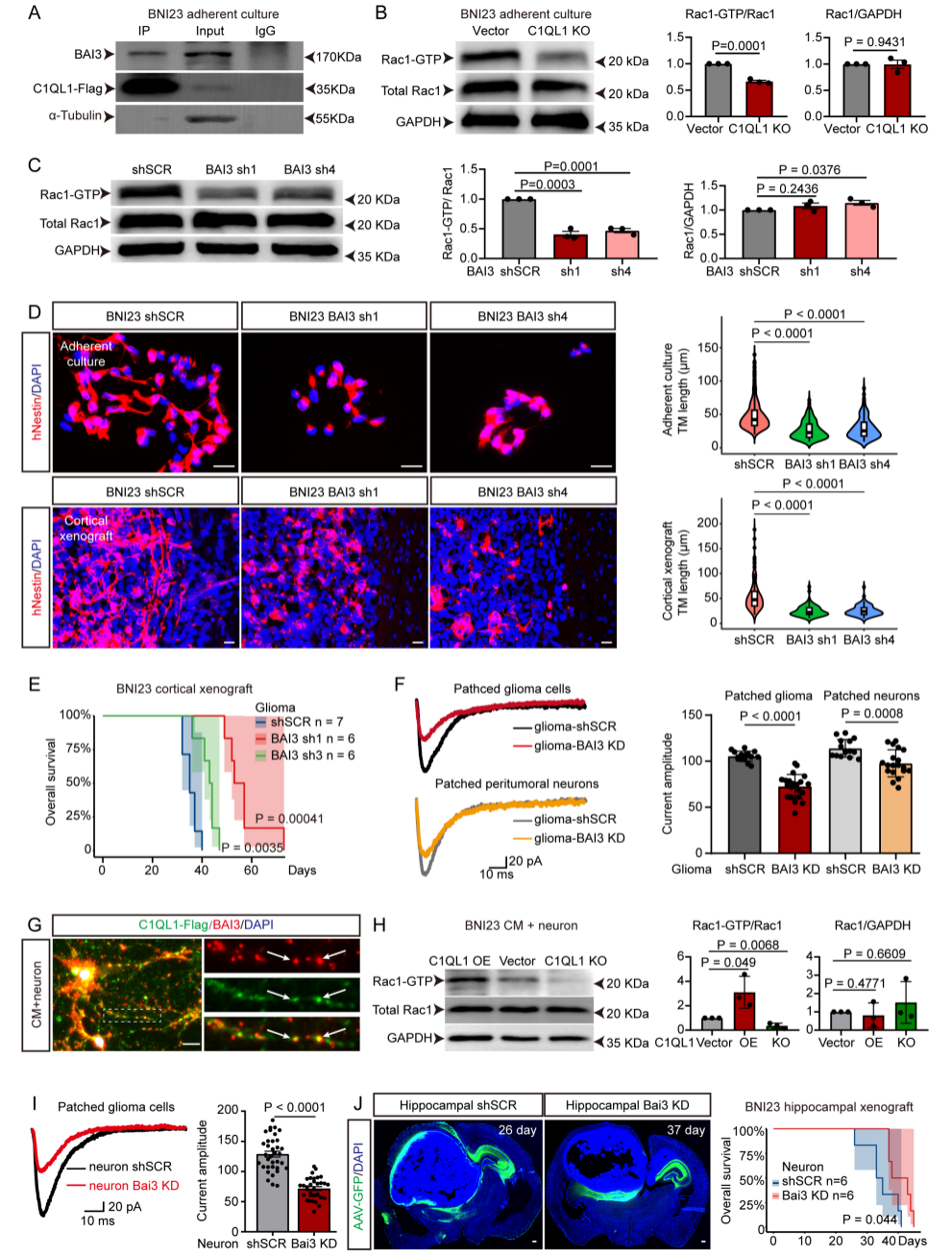
图5. C1QL1通过BAI3受体在胶质瘤和神经元中激活Rac1
鉴于C1QL1在胶质瘤细胞和神经元中均通过BAI3-Rac1通路发挥作用,作者探究靶向抑制Rac1活性是否能同时阻断其介导的胶质瘤细胞间及胶质瘤-神经元间相互作用。基于阿尔茨海默病模型中筛选的先导化合物,作者研发出首款Rac1抑制剂JK50561,其可竞争性抑制F-肌动蛋白与CORO1A/PAK1/RhoGDI复合物结合(图6A),破坏Rac1激活的细胞骨架反馈环路,作用机制区别于靶向Rac1-GEFs的传统抑制剂。JK50561可有效抑制GSC的Rac1活性(图6B),中高浓度可抑制GSC自我更新/增殖。与抑制剂1A116对比显示,高浓度(5-50 μM)时两者均能缩短GSC肿瘤微管长度(图6C、D),但低浓度(75-150 nM)时仅JK50561表现出显著抑制效果(图6C、D),抑制优势更明显。此外,JK50561可挽救GSC条件培养基诱导的神经元突触修剪(图6E),减少恶性突触数量(图6F)。综上,JK50561可破坏胶质瘤TM、挽救突触修剪并抑制恶性突触形成。
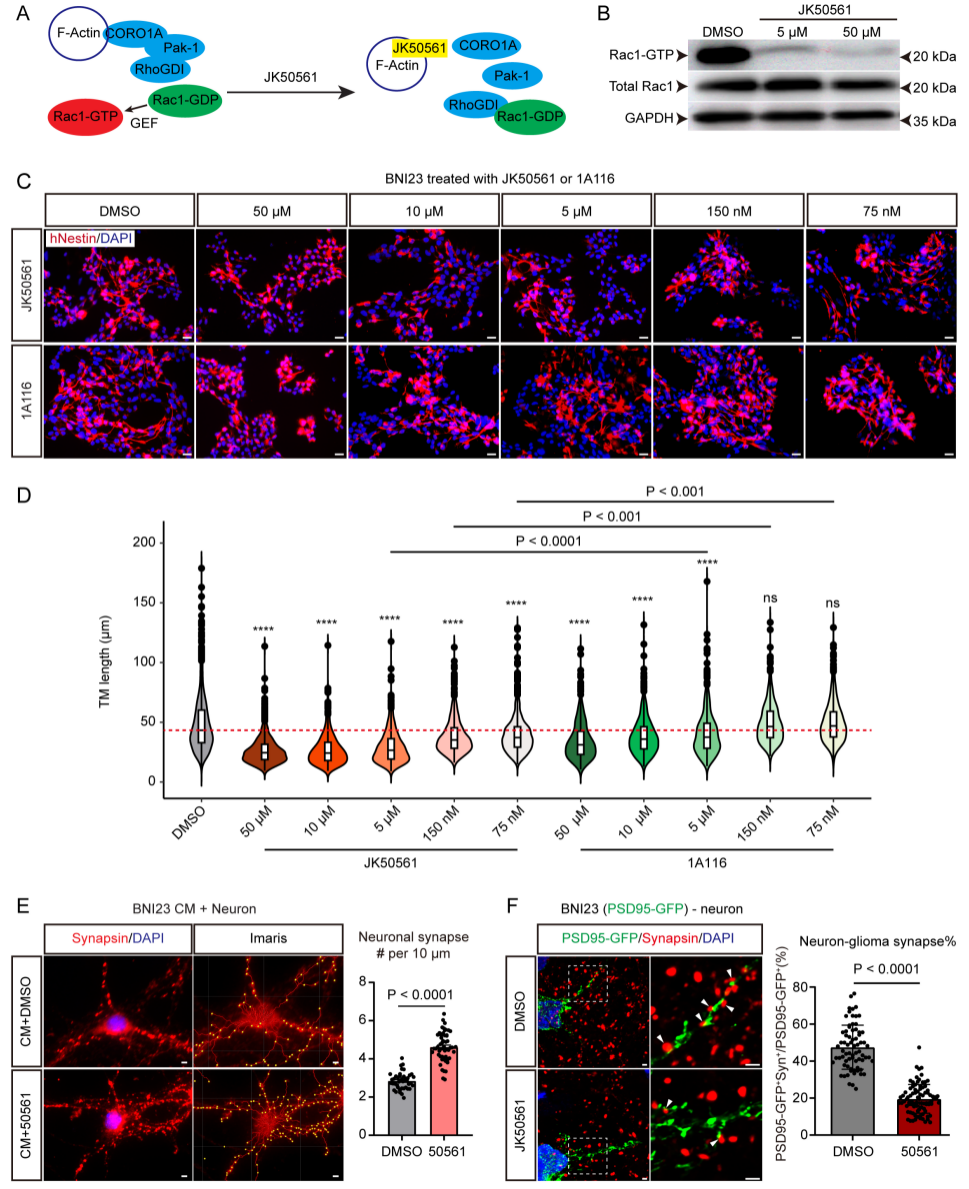
图6. Rac1抑制剂JK50561破坏TMs并挽救C1QL1诱导的神经突触修剪及恶性突触形成
为了明确JK50561对体内GBM复发的抑制作用,在GBM复发模型中开展药物治疗(图7A)。将超灵敏荧光素酶报告基因AkaLuc转染至GSC,通过纵向生物发光活体成像监测术后残留细胞复发(图7A)。GSC移植14天(dpt)后手术切除皮质肿瘤,术后第1天(dps)起每日灌胃50 mg/kg JK50561至实验终点(图7A)。结果显示,JK50561可有效抑制肿瘤生长(图7B),延长两种GSC异种移植复发模型小鼠生存期(图7C),并减小肿瘤体积、破坏hNestin阳性TM(图7D),证实其对GBM复发的抑制效果。
为了明确JK50561对体内胶质瘤细胞及相关神经元的影响,对原发肿瘤、溶媒及JK50561处理的复发肿瘤进行单核RNA测序(图7E)。溶媒处理的复发肿瘤细胞中,TM调控基因(如GAP43)及促肿瘤通路(缺氧、WNT信号等)、TM/突触相关通路均上调(图7F),而JK50561可下调上述基因及通路(图7F)。通过生物信息学方法分离肿瘤相关兴奋性神经元(ENs)(图7E),溶媒处理的复发肿瘤相关ENs中,突触/轴突发育及可塑性通路、神经元活性基因(如Fosb)均上调(图7F)。JK50561可有效降低其表达(图7F),且经FOSB染色证实,表明其通过同时影响复发胶质瘤细胞和相关神经元发挥作用。鉴于放疗是GBM标准治疗方案,对AkaLuc-GSC移植小鼠进行手术联合放疗(RT),并给予溶媒或JK50561治疗,监测肿瘤复发(图7G)。结果显示,放疗+JK50561组肿瘤生长最慢(图7G),提示两者具有协同作用。
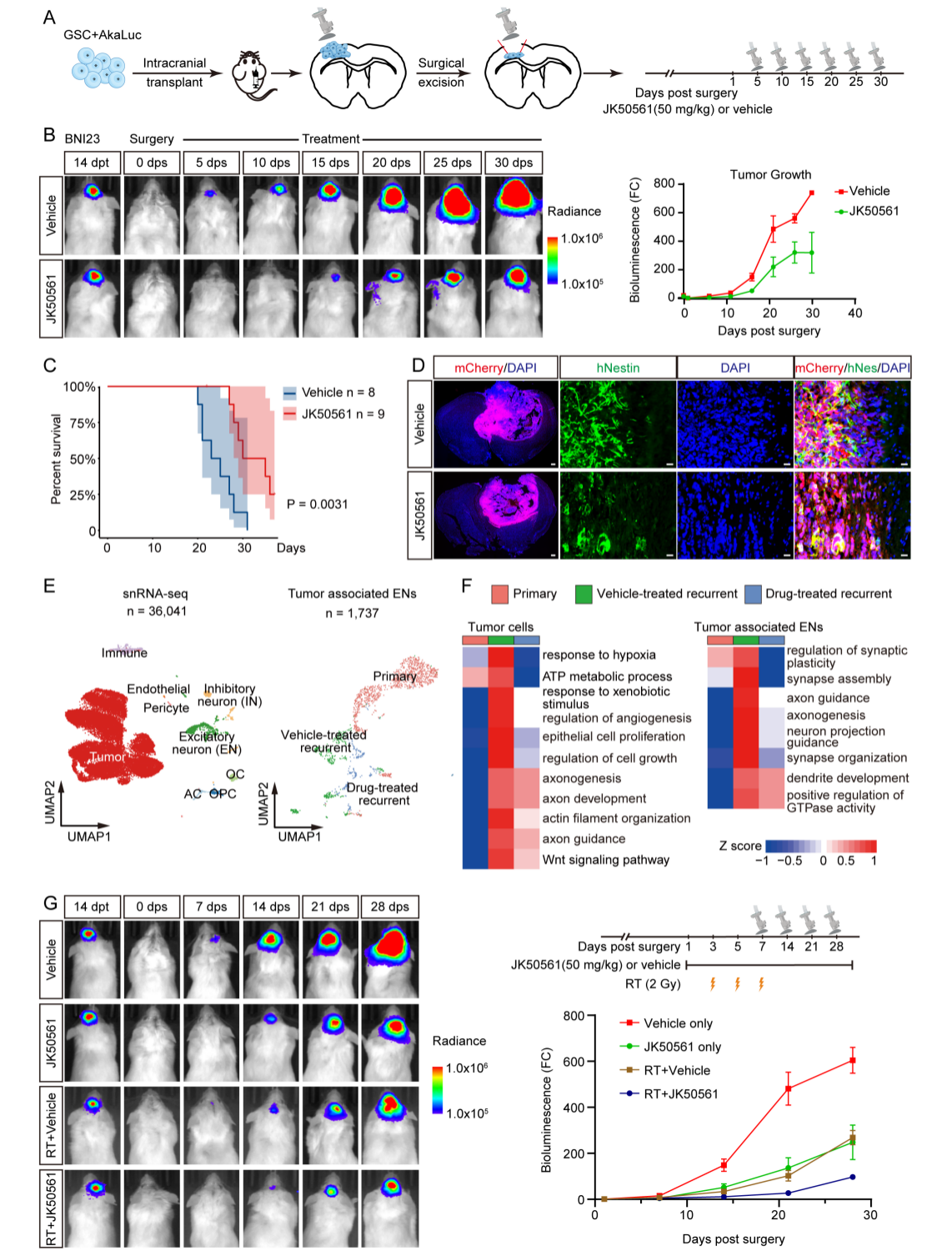
图7. JK50561治疗可延长 GBM 复发模型的生存期
本研究聚焦胶质母细胞瘤(GBM)浸润性生长与复发的核心机制,通过单细胞/空间组学、功能实验及临床模型验证,揭示了C1QL1介导的胶质瘤-神经微环境交互作用新机制,并开发了靶向该通路的新型Rac1抑制剂,为复发型GBM的治疗提供了全新策略。
本文使用的工具病毒布林凯斯均可提供:
同时布林凯斯也可提供各类定制服务请联系小布:18971216876(微信同号)或者咨询所在区域的销售经理获取更多信息。