Client Article | Sci. Transl. Med. | Xiaodong Liu & Changyu Jiang’s Team Reveals a New Mechanism of Microglial Remodeling of Spinal Pain Microcircuits—Synaptic Pruning
Release time:2025-08-15 10:18:32
The balance between excitation and inhibition in the spinal cord dorsal horn (SCDH) neural network is crucial for maintaining proper pain processing. Nerve injury disrupts this balance by suppressing inhibitory neurotransmission, leading to enhanced pain signaling. PKCγ+ interneurons, located in lamina II/III of the SCDH, play a key role in mechanical allodynia following peripheral nerve injury and persistent inflammation. Under normal conditions, these neurons are regulated by inhibitory neurons; in pathological states, this inhibition is lifted, triggering abnormal pain. Activated microglia release neuroinflammatory mediators, and recent studies have found that microglia-mediated synaptic pruning via the complement system is involved in neurological diseases. However, whether such pruning of inhibitory synapses occurs in neuropathic pain, and how it affects target cells and pain processing, has remained unclear.
On June 5, 2025, Professor Xiaodong Liu of The Chinese University of Hong Kong and Associate Researcher ChangyuJiang from the Pain Department of Shenzhen Nanshan Hospital published an article in Science Translational Medicine titled “Microglial pruning of glycinergic synapses disinhibits spinal PKCγ interneurons to drive pain hypersensitivity in mice”. The study shows that microglia in the spinal cord prune glycinergic synapses, resulting in the disinhibition of spinal PKCγ interneurons, which in turn enhances low-threshold Aβ fiber transmission and triggers pain hypersensitivity.
In Neuropathic Pain Development, SCDH Microglial Synaptic Pruning and PKCγ Interneuron Disinhibition Occur Simultaneously
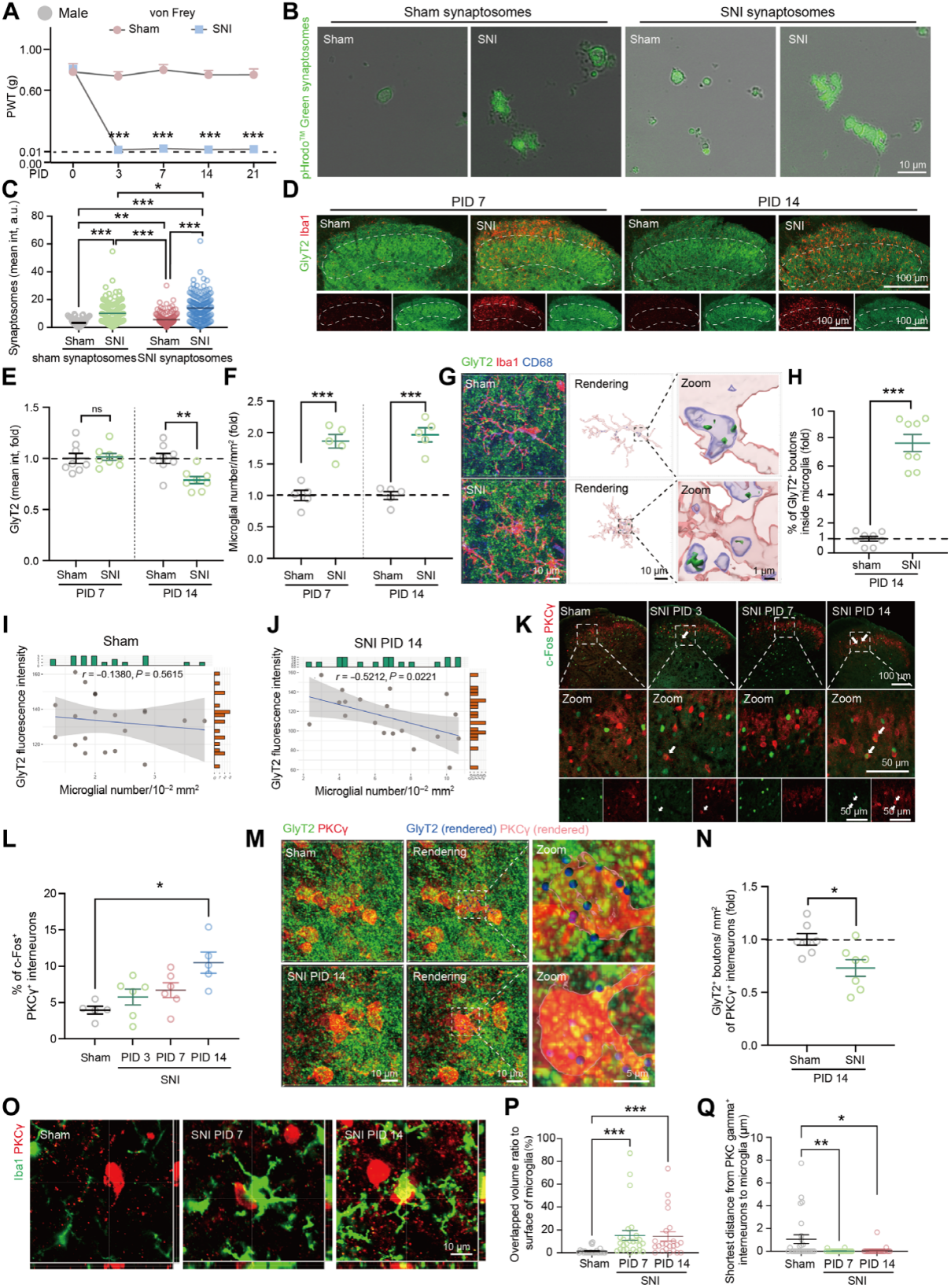
In the spared nerve injury (SNI) mouse model of neuropathic pain, mechanical hypersensitivity appeared after injury (Fig. 1A). By postoperative day 3 (PID3), the number of Iba1-positive microglia in the ipsilateral SCDH significantly increased, remaining elevated through PID14; lysosome-associated protein CD68 levels in microglia stayed high for at least 21 days. In vitro assays confirmed that at PID14, microglia from SNI mice had significantly higher phagocytic capacity than sham controls, engulfing more synaptic boutons (Fig. 1B–C).
Analysis of synaptic markers showed that, compared with the sham group, inhibitory synapse presynaptic terminal density (GlyT2 for glycinergic neurons; GAD65/67 for GABAergic neurons) decreased at PID14 in SNI mice, accompanied by increased microglial density. In contrast, excitatory synapses (VGluT1, VGluT2, Homer1—markers for glutamatergic neurons) remained unchanged (Fig. 1D–F). The density of glycinergic neuron somata did not change, although expression of related genes increased; inhibitory neuron (Pax2+) density was also unchanged.
Phagocytosis analysis showed that, at PID14, the number of inhibitory presynaptic elements within microglial lysosomes increased significantly—glycinergic and GABAergic presynaptic components increased 7.7-fold and 1.8-fold, respectively (Fig. 1G–H). In SNI mice, microglial density in lamina II–III of the SCDH negatively correlated with GlyT2 immunoreactivity (Fig. 1I–J). Under dynamic brush stimulation (pain response test), the proportion of c-Fos+/PKCγ+ interneurons was higher in SNI mice than in sham controls at PID14 (Fig. 1K–L). At PID14, the number of glycinergic terminals on PKCγ+ interneurons in SNI mice was lower than in sham mice, while no difference was observed at PID7 (Fig. 1M–N). At both PID7 and PID14, activated microglia were observed surrounding PKCγ+ interneuron somata (Fig. 1O–Q).
The same results were validated in female mice, including pain hypersensitivity, microglial activation, loss of inhibitory synapses, and PKCγ+ neuron activation. In summary, microglial phagocytosis of glycinergic presynaptic elements and PKCγ+ interneuron disinhibition jointly drive the development of neuropathic pain.
Fig. 1 Enhanced microglial phagocytosis of inhibitory presynaptic components and spinal disinhibition in male mice after peripheral nerve injury.
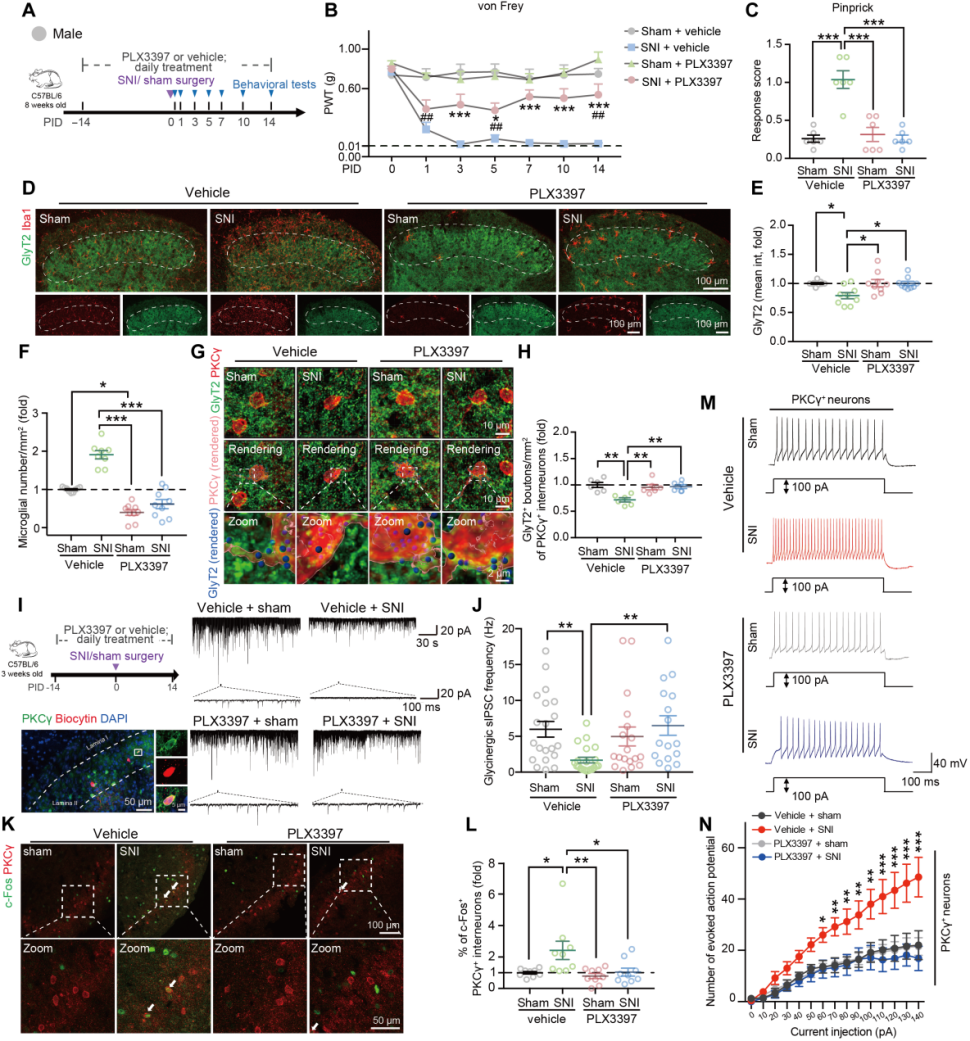
To determine whether microglia play a key role in central disinhibition after SNI, the colony-stimulating factor 1 receptor (CSF1R) inhibitor PLX3397 was administered orally from 14 days before sham/SNI surgery to deplete myeloid cells (mainly microglia) in the SCDH (Fig. 2A). The results showed that microglial depletion alleviated SNI-induced mechanical hypersensitivity (Fig. 2B–C), reversed the loss of glycinergic presynaptic terminals at PID14 (Fig. 2D–E), eliminated SNI-induced microglial activation (Fig. 2D, F), and reduced the decrease in GlyT2-positive puncta on PKCγ+ interneurons (Fig. 2G–H). Further examination revealed that in vivo depletion of microglia eliminated the SNI-induced reduction in glycinergic sIPSC frequency in PKCγ+ interneurons (Fig. 2I–J). Compared with the sham group, SNI increased the excitability of PKCγ+ interneurons, whereas microglial depletion abolished this hyperexcitability (Fig. 2K–N) without affecting the sham group.
Fig. 2 Microglial depletion alleviates glycinergic synapse loss, PKCγ+ interneuron hyperexcitability, and mechanical allodynia induced by SNI in male mice.
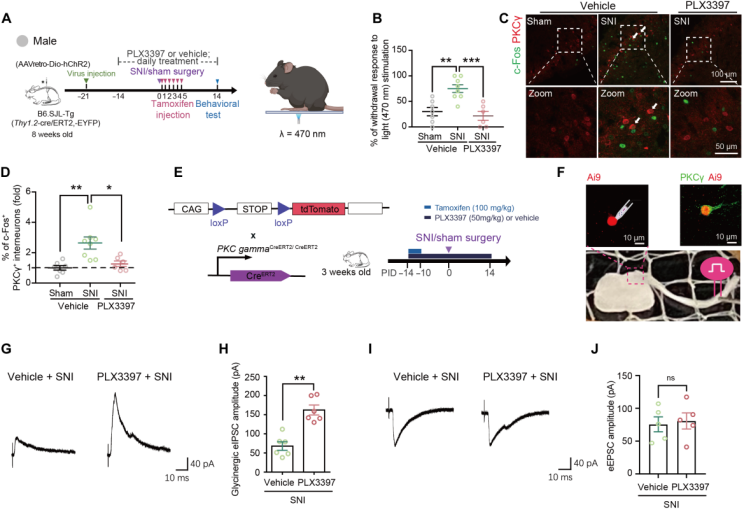
To investigate whether microglial depletion could reverse SNI-induced abnormal pain, optogenetic activation of Aβ fibers was used to assess pain-like behavior. In Thy1.2-cre/ERT2-EYFP mice, ChR2 was expressed in large-diameter neurons by injecting RetroAAV-hSyn-DIO-ChR2-mCherry into the left L4–L6 spinal dorsal horn, enabling ChR2 expression in both somata and peripheral skin terminals (Fig. 3A), thereby specifically targeting Aβ low-threshold mechanoreceptors (LTMRs). At PID14, blue light was applied to the lateral plantar surface of the ipsilateral hind paw to stimulate Aβ fibers (Fig. 3A). Optogenetic activation of Aβ fibers induced a higher paw withdrawal rate in SNI mice than in sham-operated mice (Fig. 3B), indicating the presence of tactile allodynia. Microglial depletion suppressed the responsiveness of SNI mice to Aβ fiber activation (Fig. 3B). In addition, Aβ fiber activation in SNI mice induced a higher proportion of c-Fos+/PKCγ+ interneurons and increased the density of c-Fos+ cells in lamina I; microglial depletion reduced these proportions and densities (Fig. 3C–D).
To examine whether microglial depletion regulates the responsiveness of PKCγ+ interneurons to low-threshold Aβ primary afferent inputs, whole-cell patch-clamp recordings were performed on tdTomato-expressing neurons in spinal cord slices from PKCγ-CreERT2:Ai9 mice (Fig. 3E–F). Recordings showed that PLX3397 treatment increased the amplitude of glycinergic eIPSCs in tdTomato neurons from SNI mice (Fig. 3G–H) but did not affect the amplitude of Aβ fiber–evoked eEPSCs (Fig. 3I–J).
Furthermore, intrathecal administration of minocycline, which inhibits spinal microglial activation, alleviated pain sensitivity, reversed GlyT2+ terminal loss, and suppressed activation of related neurons and LPBN cells. Control experiments (splenectomy, intrathecal saline injection) ruled out procedural artifacts and peripheral immune cell infiltration effects. In summary, blocking microglia-mediated phagocytosis of inhibitory synapses partially restored the structure and function of spinal inhibitory circuits.
Fig. 3 Microglial depletion suppresses SNI-induced enhancement of behavioral and neuronal responses to Aβ fiber activation.
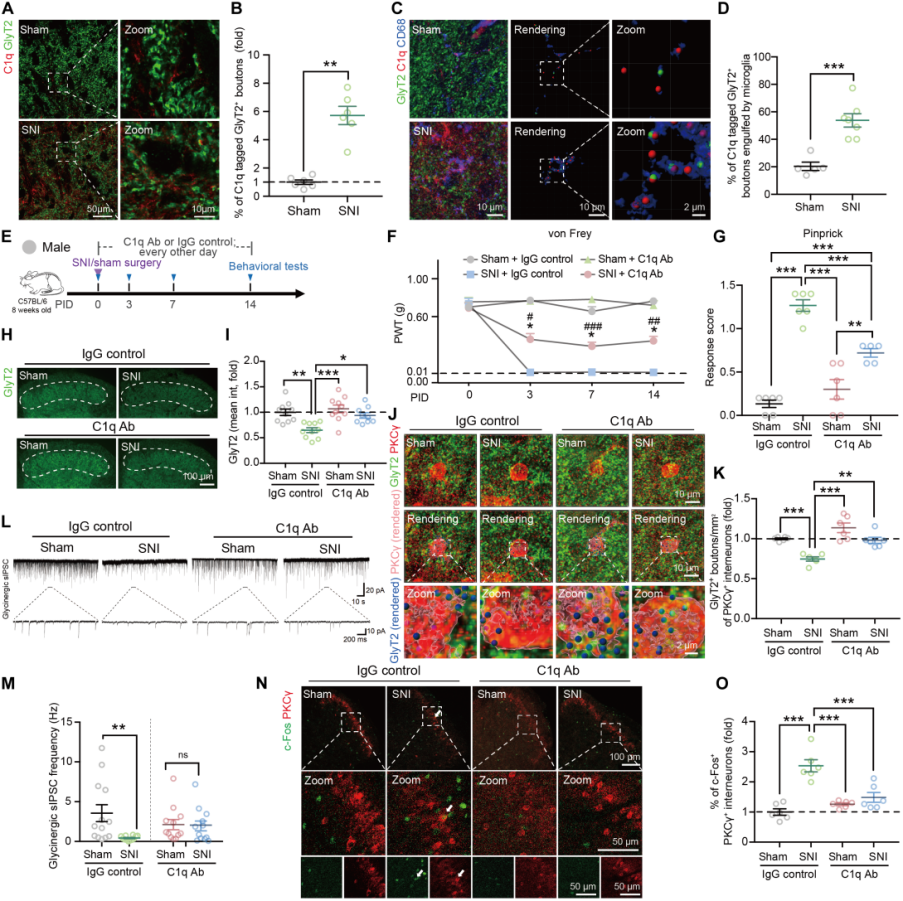
The complement system influences synaptic pruning by tagging synapses and sending “eat me” or “don’t eat me” signals. After SNI, C1q expression increased in both the SCDH and isolated SCDH microglia of male and female mice. In addition, compared with sham controls, C1q immunoreactivity colocalized with glycinergic presynaptic boutons in the SCDH of SNI mice was approximately sixfold higher (Fig. 4A–B), and a greater proportion of C1q-labeled GlyT2+ puncta were engulfed by microglia (Fig. 4C–D).
Blocking C1q with a neutralizing antibody (Fig. 4E) partially alleviated mechanical hypersensitivity on day 14 after SNI (Fig. 4F–G); reversed the loss of glycinergic presynaptic terminals (Fig. 4H–I); restored the density of presynaptic boutons associated with PKCγ+ interneurons (Fig. 4J–K) as well as the frequency and amplitude of sIPSCs (Fig. 4L–M). Moreover, anti-C1q antibody reduced the proportion of c-Fos+/PKCγ+ interneurons in SNI mice (Fig. 4N–O).
Fig. 4 SNI-induced glycinergic synaptic pruning and mechanical allodynia both depend on the C1q signaling pathway.
Single-Cell Sequencing Identifies APOE⁺ Microglia
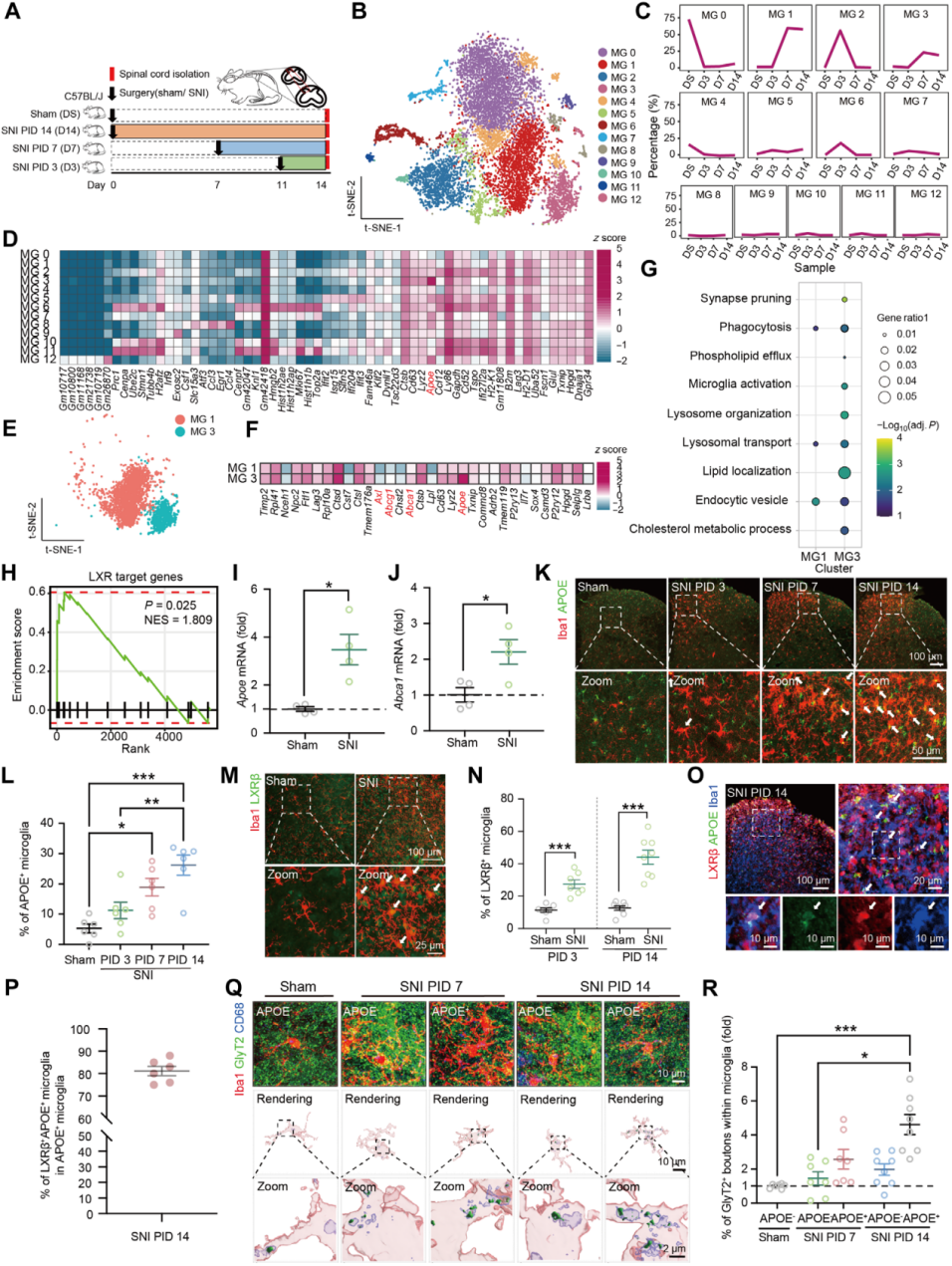
Using a single-cell RNA sequencing platform, transcriptomic analysis was performed on myeloid cells sorted by FACS from the ipsilateral SCDH of SNI or sham-operated mice. At PID3, 7, and 14, tissue from the ipsilateral L4–L6 SCDH was collected (Fig. 5A), and Cd11b⁺ cells were isolated using the monocyte/macrophage surface marker Cd11b for fluorescence-activated cell sorting, followed by sequencing. Microglia were classified into 16 clusters (MG0–15), with three minor clusters excluded (Fig. 5B).
Differentially expressed gene (DEG) analysis for each cluster (Fig. 5D) revealed that MG1 and MG3 emerged at PID7 and persisted through PID14 (Fig. 5C). MG1 showed low expression of homeostatic and proliferation-related genes, but high expression of H2-D1 and B2m (Fig. 5D). MG3 exhibited low expression of metabolic and homeostatic genes, but high expression of Apoe, Lyz2, and Cd63 (Fig. 5D). GO enrichment analysis of MG1 and MG3 DEGs from SNI-PID7 and PID14 samples (Fig. 5E–G) showed that MG3 DEGs were significantly enriched in synaptic pruning pathways (Fig. 5G), as well as in phagocytosis-related genes (Axl) and liver X receptor (LXR) target genes such as Apoe, Abca1, and Abcg1 (Fig. 5F, H).
qRT-PCR and immunofluorescence staining confirmed that Apoe expression increased in the SCDH of both male and female mice at PID7 and PID14 after SNI, with a higher proportion of APOE⁺ microglia (Fig. 5I–L). Nearly all APOE⁺ microglia colocalized with LXR (Fig. 5M–P) and contained more glycinergic components than APOE⁻ cells (Fig. 5Q–R). Furthermore, microglia without APOE expression at PID14 still exhibited an activated phenotype, indicating that APOE is not a universal marker of activation, and that the MG3 transcriptional profile resembles that of disease-associated microglia (DAM).
Fig. 5 Microglia expressing LXR/APOE are upregulated after sciatic nerve injury and engulf glycinergic synapses.
LXR Enhances APOE Expression in Microglia, Which Is Essential for Glycinergic Synapse Pruning and SNI-Induced Mechanical Hypersensitivity
Apoe is a known downstream transcriptional target of LXR. To investigate whether the LXR–APOE axis is involved in microglia-mediated loss of inhibitory synapses in the SCDH during neuropathic pain, immortalized microglial (IMG) cells were treated with the LXR agonist GW3965. GW3965 upregulated the expression of Apoe, Abca1, and Abcg1, and enhanced phagocytic activity. These genes are among those upregulated in the APOE-dominant microglial subpopulation identified in the single-cell RNA-seq dataset.
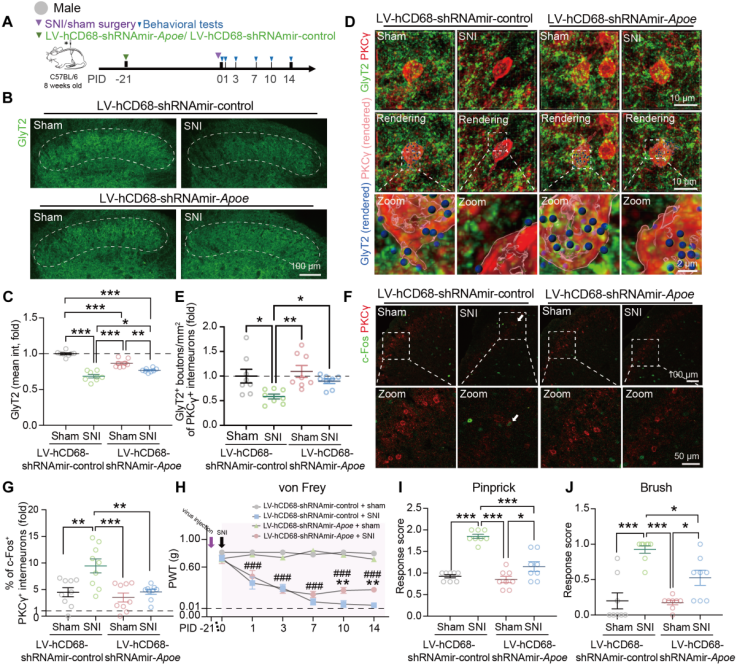
A lentiviral vector (LV-hCD68-shRNAmir-Apoe) was constructed to achieve microglia-specific Apoe knockdown. In vitro, Apoe knockdown abolished the GW3965-induced increase in phagocytic activity. In vivo, injection of Apoe-targeting lentivirus reduced Apoe expression in SCDH microglia (Fig. 6A), decreased the proportion of APOE⁺ microglia, alleviated SNI-induced loss of glycinergic presynaptic elements, reduced anchoring to PKCγ⁺ interneurons (Fig. 6B–E), and diminished the increase in c-Fos⁺/PKCγ⁺ cells (Fig. 6F–G).
To determine whether microglial APOE is involved in maintaining pain hypersensitivity, Apoe knockdown was performed at different time points. Knockdown before or in the early stages of SNI reduced hypersensitivity, with similar results observed in female mice. Experiments in global Apoe knockout mice showed that Apoe deletion abolished the SNI-induced increase in microglial synaptosome phagocytosis, reversed glycinergic synapse loss, and suppressed PKCγ⁺ interneuron activation. Pain-like behaviors were assessed in SCDH microglia-specific Apoe knockdown mice (Fig. 6H–J) and in Apoe-deficient mice. Compared with wild-type or littermate controls, both Apoe deficiency and microglia-specific Apoe knockdown reduced SNI-induced dynamic allodynia, pinprick hypersensitivity, and mechanical allodynia.
Finally, intrathecal injection of CSF1 together with GW3965 increased the proportion of APOE⁺ microglia in the SCDH, leading to more CD68⁺ lysosomes in microglia, decreased GlyT2⁺ inhibitory synapse density, and increased GlyT2⁺ remnants within lysosomes of activated microglia.
Fig. 6 Deletion of microglial APOE inhibits SNI-induced glycinergic synapse pruning and alleviates mechanical hypersensitivity.
LXR Blockade or Apoe Deficiency Inhibits Microglial C1q Production
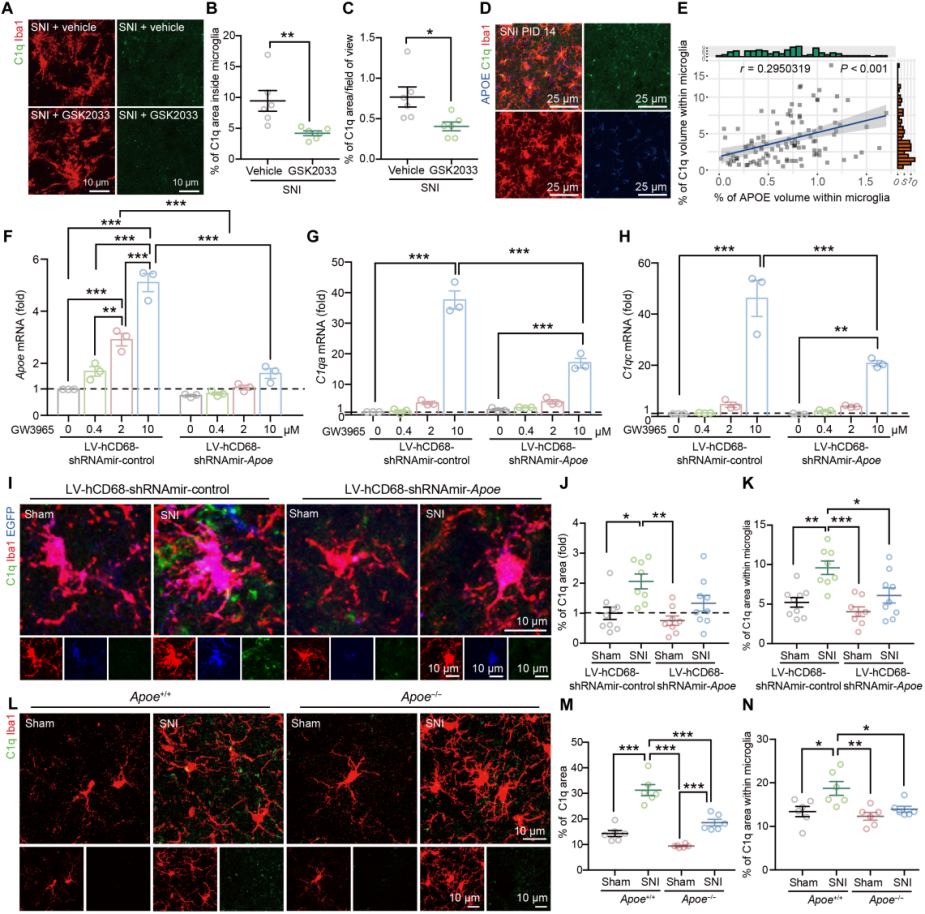
To explore whether the LXR–APOE axis regulates C1q production in microglia, immortalized microglial (IMG) cells were treated with the LXR agonist GW3965 or the antagonist GSK2033. The expression levels of C1q, Apoe, Abca1, and Abcg1 changed correspondingly. In mice co-treated with CSF1 and GW3965, C1q expression in the SCDH increased, whereas no such change was observed in control groups. Furthermore, GSK2033 suppressed the SNI-induced increase in C1q density within SCDH microglia (Fig. 7A–C).
Given that C1q expression in SNI mice positively correlated with the density of APOE⁺ microglia (Fig. 7D–E), the role of APOE in C1q regulation was further examined. Knockdown of Apoe in IMG cells reduced GW3965-induced expression of C1q-related genes (Fig. 7F–H), and primary microglia from Apoe⁻/⁻ mice displayed altered C1q-related gene expression. Both microglia-specific Apoe knockdown mice and Apoe⁻/⁻ mice showed reduced SNI-induced C1q production in spinal microglia (Fig. 7I–N).
Fig. 7 The microglial LXR–APOE axis regulates complement activation.
Conclusion
Using a mouse SNI model, this study demonstrates that spinal microglia promote disinhibition of PKCγ⁺ interneurons by pruning glycinergic synapses, thereby facilitating low-threshold Aβ-fiber transmission and inducing mechanical hypersensitivity. Highly expressed Apoe and complement C1q in microglia act as key mediators in this process. Deletion of Apoe, microglia-specific knockdown of Apoe, or administration of anti-C1q antibody can reverse these changes and alleviate hypersensitivity. These findings reveal a mechanistic role for microglia-mediated pruning of inhibitory synapses in the development of neuropathic pain and provide potential therapeutic targets and strategies aimed at restoring inhibitory gating to control pain hypersensitivity.
Viral vectors used in this study are available from BrainCase
Service Type :
Select the service you'd like to purchase.
Order Information(Premade-AAVs)
Please provide us some information about the service you'd like to order.
Order Information(Custom AAV/Lentivirus)
Please provide us some information about the service you'd like to order.
Order Information(Others)
Please provide us some information about the service you'd like to order.