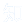抑郁症的发病涉及遗传与环境双重因素,患者常表现为持续的情绪低落与兴趣丧失,部分存在治疗抵抗。慢性应激是其重要诱因,可过度激活下丘脑-垂体-肾上腺(HPA)轴,导致皮质酮水平升高,损害海马等情绪调控脑区功能;其中海马脑内腺苷酸环化酶(ADCYs)及下游cAMP信号通路的下调可能与抑郁发生有关,但具体机制尚未完全阐明。
2025年12月12日,吉林大学孙栋团队、东北师范大学朱筱娟团队及广州医科大学陈文兵团队联合在Advanced Science发表题为“Adenylyl cyclase 8 in dorsal CA1 neurons prevents depressive-like behaviors by maintaining neuronal excitability and glutamatergic neurotransmission through TIP39-PTH2R signaling”的研究论文。该研究表明背侧海马体(dCA1)神经元中腺苷酸环化酶8(ADCY8)的功能障碍是抑郁症发生发展的关键风险因素。慢性应激选择性降低该脑区Adcy8表达,其缺失通过丝裂原活化蛋白激酶(MAPK)信号通路下调甲状旁腺激素2受体(PTH2R),进而损害神经元兴奋性与谷氨酸能传递,最终诱发抑郁样行为。

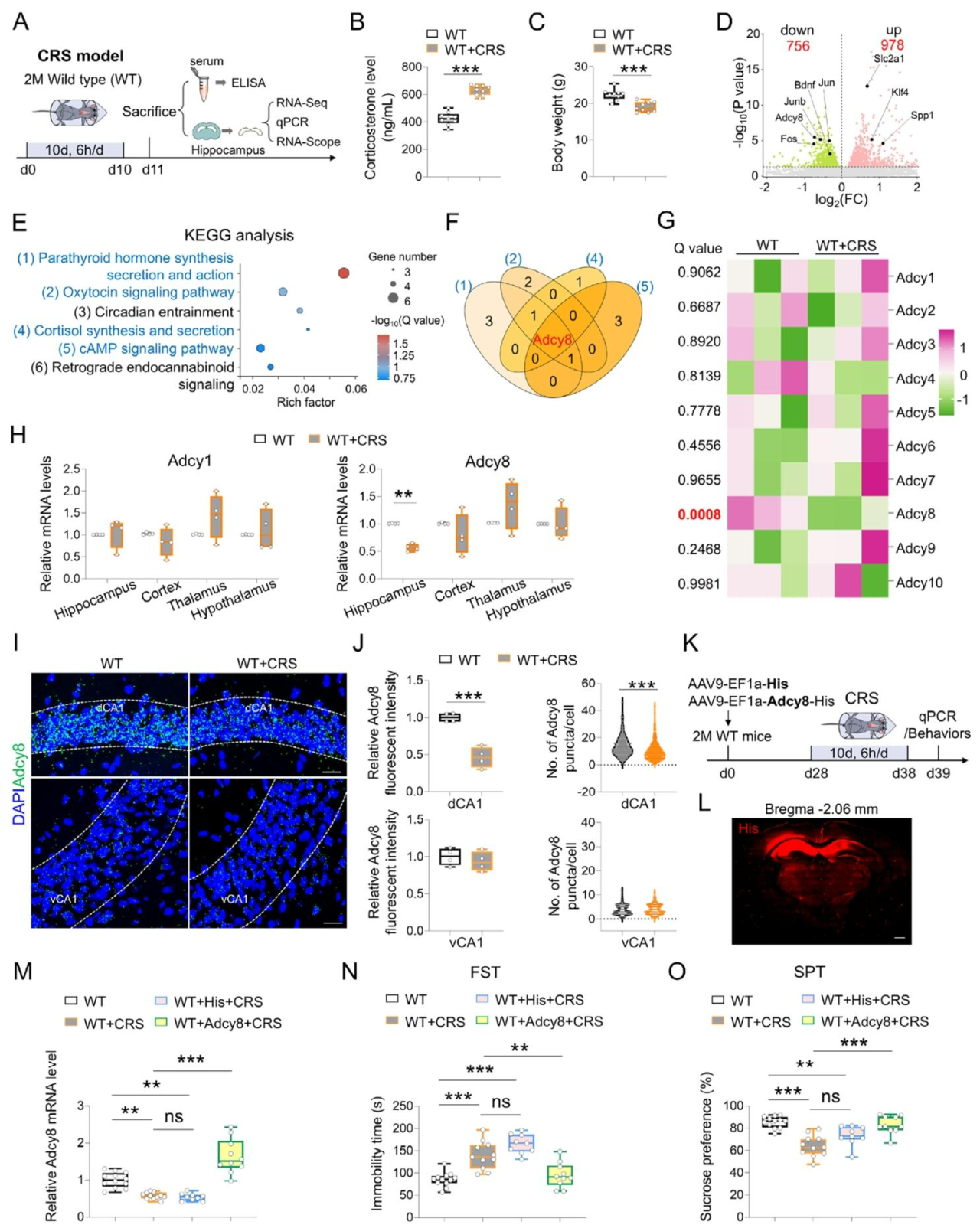
图1 CRS选择性降低海马体中Adcy8的表达,且上调Adcy8表达可逆转CRS小鼠的抑郁样行为。
为探究海马Adcy8在情绪行为调控中的作用,RNA-seq发现小鼠海马Adcy8主要表达于CA锥体细胞、DG颗粒细胞、GABA能神经元并在CA1锥体细胞、DG 颗粒细胞及 PV⁺中间神经元富集,胶质细胞中几乎无表达(图2A-C)。RNA 原位杂交显示Adcy8在 dCA1 高表达、dDG 中度表达、dCA3 极低表达,vCA1 表达显著低于dCA1,vCA3及vDG表达与背侧对应脑区相当(图2D-E)。Adcy8探针与细胞标志物共染色证实Adcy8在dCA1区CaMKII⁺锥体细胞和PV⁺中间神经元高表达,其余胶质/小胶质细胞中低表达或无表达(图2F-G)。
图2 Adcy8在海马dCA1神经元中高表达
为明确Adcy8缺陷与小鼠神经精神疾病的关联,构建全身Adcy8敲除(KO)小鼠(图3A),海马RNA-scope证实KO小鼠Adcy8 mRNA几乎缺失(图3B);且KO小鼠FST不动时间延长、SPT偏好率降低(图3C、D),提示Adcy8调控抑郁样及焦虑样行为。为明确组织/细胞特异性功能,构建Adcy8CaMKII-CKO(兴奋性神经元敲除)和Adcy8PV-CKO(抑制性神经元敲除)小鼠(图3E),RNA-scope验证Adcy8CaMKII-CKO小鼠dCA1神经元Adcy8 mRNA缺失(图3F);行为学显示其仅FST、SPT呈抑郁样行为(图3G、H),而Adcy8PV-CKO小鼠无显著差异,表明Adcy8通过CaMKII-Cre⁺兴奋性神经元调控抑郁样行为。进一步向Adcy8f/f小鼠dCA1注射CaMKII-Cre病毒(图3I),证实dCA1敲除Adcy8可降低mRNA水平(图3J)并重现抑郁样行为(图3K、L),而vCA1敲除无此效应(图3M-P),最终证实dCA1兴奋性神经元中Adcy8敲除致小鼠抑郁样行为。
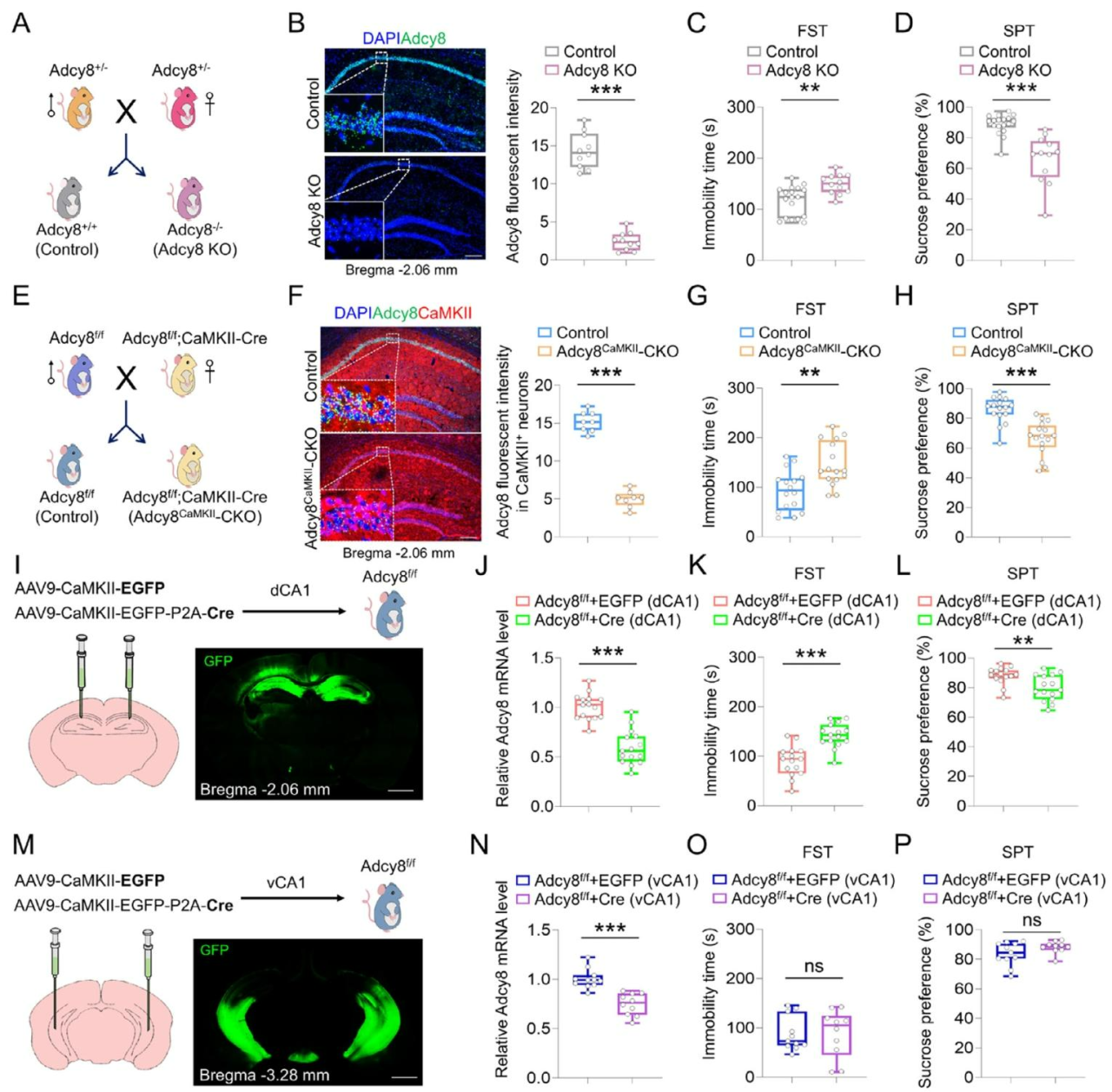
图3 dCA1神经元中Adcy8条件性敲除(CKO)导致小鼠出现抑郁样行为
进一步向dCA1区注射AAV9-CaMKII-GCaMP6m后记录发现(4D、E),在自发活动状态下,两组小鼠的钙信号无显著差异(图4F、G);当施加ARS或FST挣扎行为可迅速引发对照小鼠dCA1神经元钙信号升高,而Adcy8缺失小鼠则无此响应(图4H-M)。综上,Adcy8为应激诱导dCA1神经元钙信号激活所必需,其缺失可能导致神经元应激反应性降低。
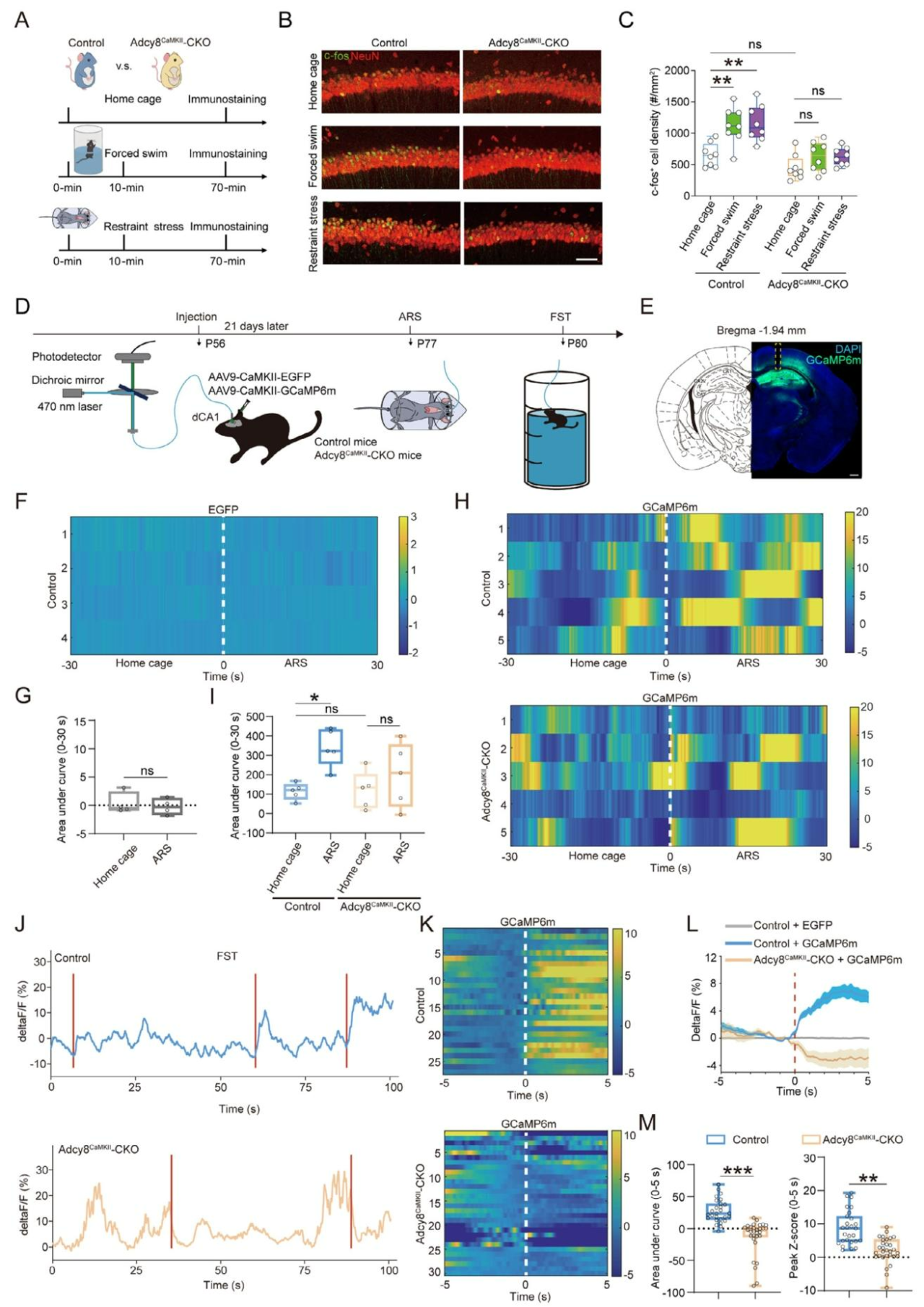
图4 Adcy8CaMKII-CKO小鼠海马体神经元活性降低
为探究Adcy8影响dCA1神经元功能的机制,研究对其进行了电生理与形态学分析(图5A)。膜片钳记录显示,Adcy8CaMKII-CKO小鼠dCA1神经元在静息膜电位无明显改变(图5B),在去极化刺激下放电频率显著降低(图5C、D),提示该小鼠dCA1神经元内在兴奋性降低。突触功能分析进一步发现,敲除小鼠的自发性兴奋性突触后电流(sEPSC)频率下降(图5E-G),而自发性抑制性突触后电流(sIPSC)无变化(图5H-J),提示谷氨酸能传递特异性受损。Adcy8CaMKII-CKO小鼠的配对脉冲比率(PPR值)与对照小鼠相当(图5K、L),排除了小鼠存在突触前释放缺陷的可能性。
将Adcy8f/f;Thy1-GFP小鼠与Adcy8CaMKII-CKO小鼠杂交,构建对照;GFP小鼠和Adcy8CaMKII-CKO;GFP小鼠,借助Thy1启动子驱动GFP信号标记兴奋性神经元。结合神经元形态特征分析,Adcy8敲除导致dCA1神经元树突总长度、分支数量及复杂性显著降低,成熟树突棘数量减少,(尤其是作为突触后位点的蘑菇状成熟棘突)(图5M-Q)。这些结果表明,综上,Adcy8缺失可损害dCA1神经元的电生理兴奋性、谷氨酸能突触传递及树突形态,从多维度揭示了其调控神经元功能的具体机制。
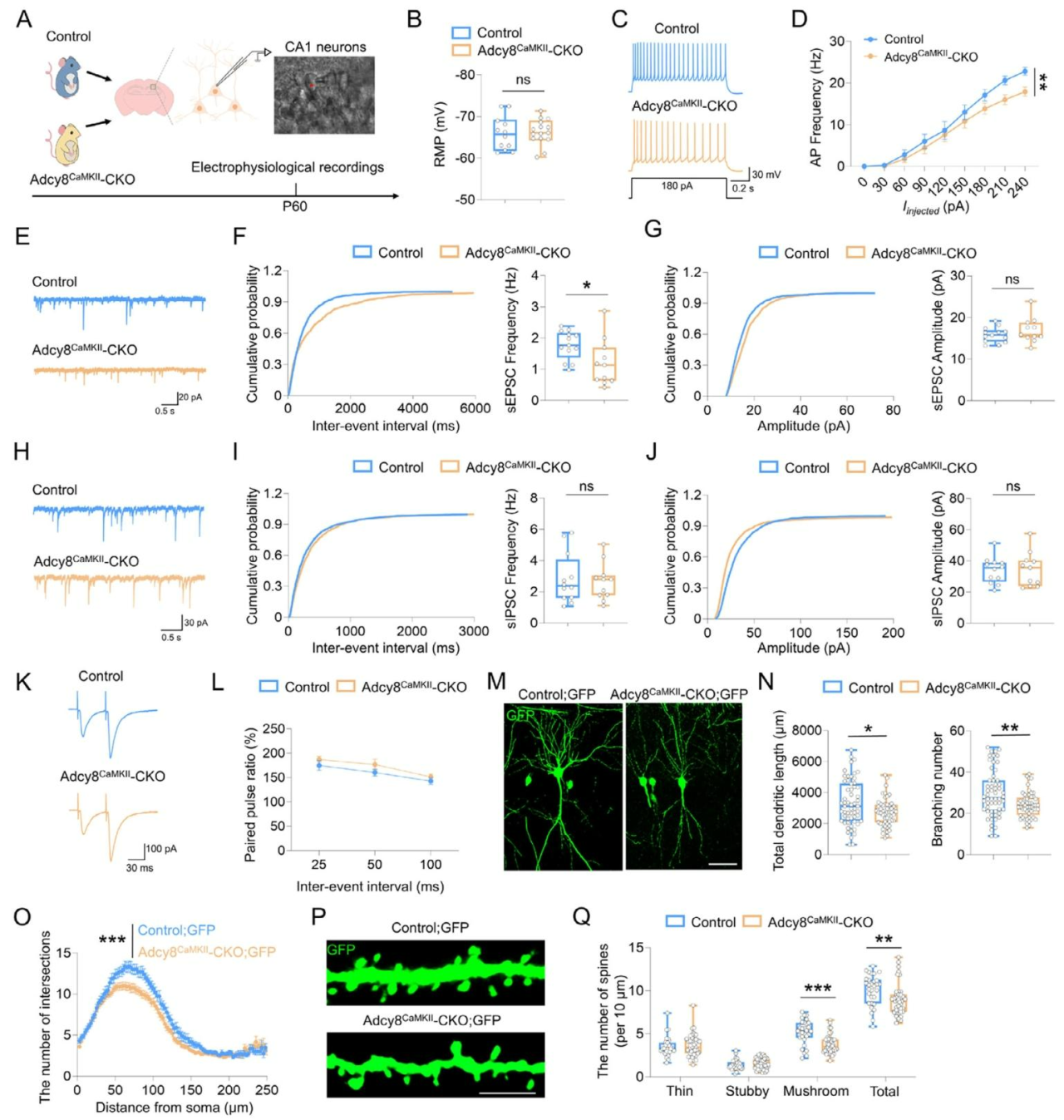
图5 Adcy8CaMKII-CKO小鼠dCA1神经元的电生理特性与形态学受损。
为深入探究Adcy8调控抑郁样行为与突触功能的分子机制,研究对Adcy8CaMKII-CKO小鼠海马组织进行RNA测序分析。结果显示,Adcy8敲除后海马基因表达谱发生显著改变,其中MAPK信号通路相关基因集显著下调(图6A-D)。蛋白水平验证进一步表明,Adcy8CaMKII-CKO小鼠海马中p-PKA、p-MEK、p-ERK1/2等MAPK通路关键蛋白磷酸化水平降低(图6E、F)。GO富集分析发现差异表达基因在甲状旁腺激素受体活性等方面显著富集;进一步筛选显示,甲状旁腺激素2受体(PTH2R)编码基因Pth2r在敲除小鼠中表达显著下降(图6G)。Westernblot及免疫染色证实Adcy8CaMKII-CKO小鼠海马PTH2R蛋白水平降低(图6H-K),这一变化与慢性应激小鼠中的表型一致。为验证MAPK信号下调是否介导PTH2R减少及行为表型,研究给予Adcy8CaMKII-CKO小鼠ERK1/2选择性激动剂PA干预后,小鼠海马p-ERK1/2及PTH2R蛋白水平均得以恢复(图6M、N),且抑郁样行为显著改善(图6O、P)。综上,Adcy8可能通过cAMP-MAPK信号轴调控PTH2R表达,进而影响抑郁样行为,PTH2R是该通路中关键的下游效应分子(图6Q)。
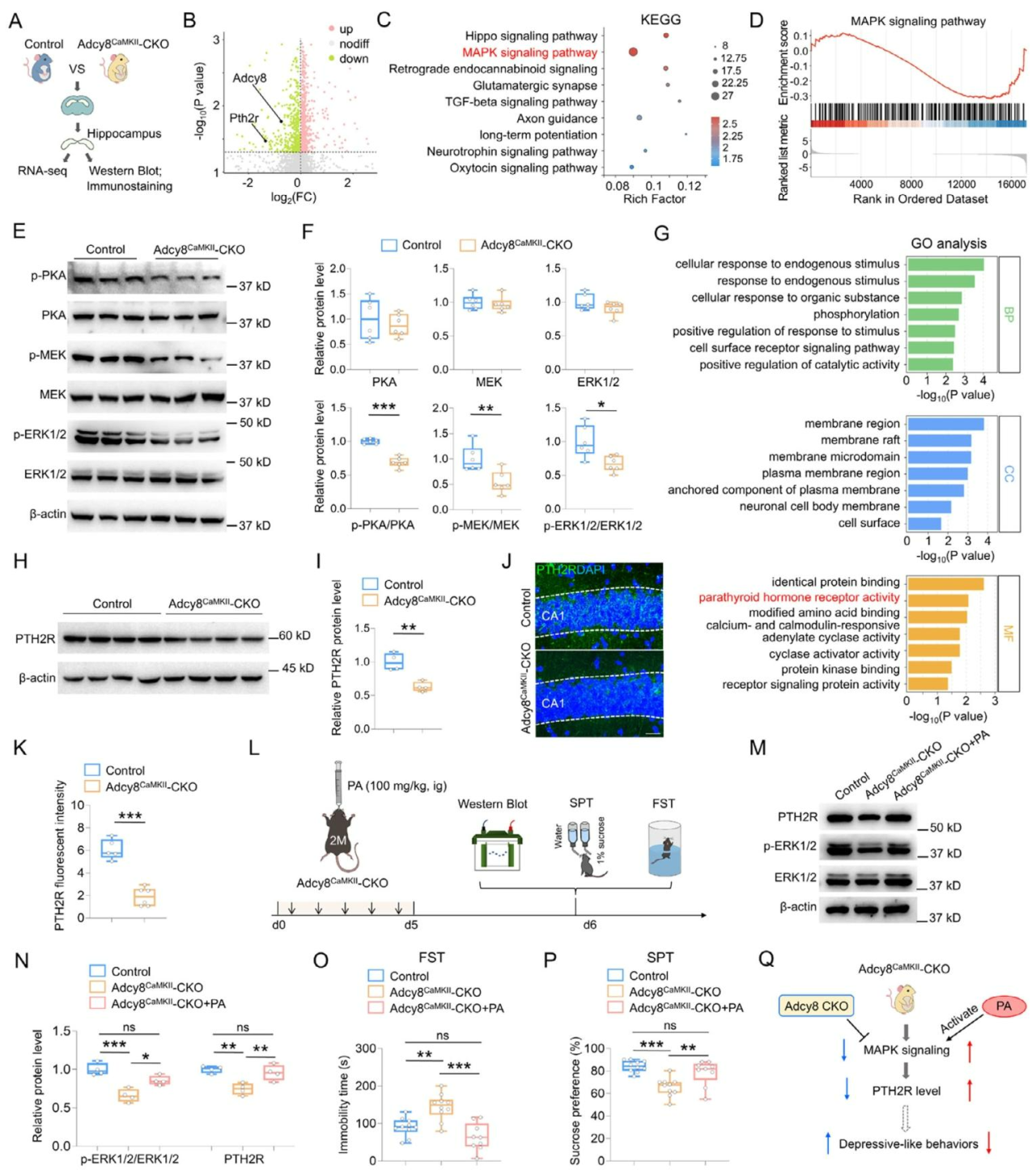
图6 Adcy8CaMKII-CKO小鼠中,MAPK信号通路受抑制且PTH2R水平降低。
为验证PTH2R对dCA1神经元电生理及抑郁样行为的调控作用,研究通过在WT小鼠双侧dCA1区注射CaMKII启动子表达Pth2r-shRNA腺相关病毒(AAV)(Pth2r-shRNA组)(图7A)。3周后,Westernblot证实PTH2R蛋白水平显著下降(图7B、C);电生理记录显示,Pth2r-shRNA组小鼠的神经元放电频率降低(图7D、E),sEPSC频率下降而sIPSC无变化(图7F-K),且PPR未受影响(图7L、M),表明其可特异性损害神经元兴奋性与谷氨酸能传递。行为学实验中,Pth2r-shRNA组小鼠在FST中不动时间延长、SPT中蔗糖偏好率下降(图7N、O),表现出抑郁样行为。综上,dCA1区Pth2r敲低可重现Adcy8缺失引起的电生理与行为表型,PTH2R是Adcy8下游的关键效应分子。
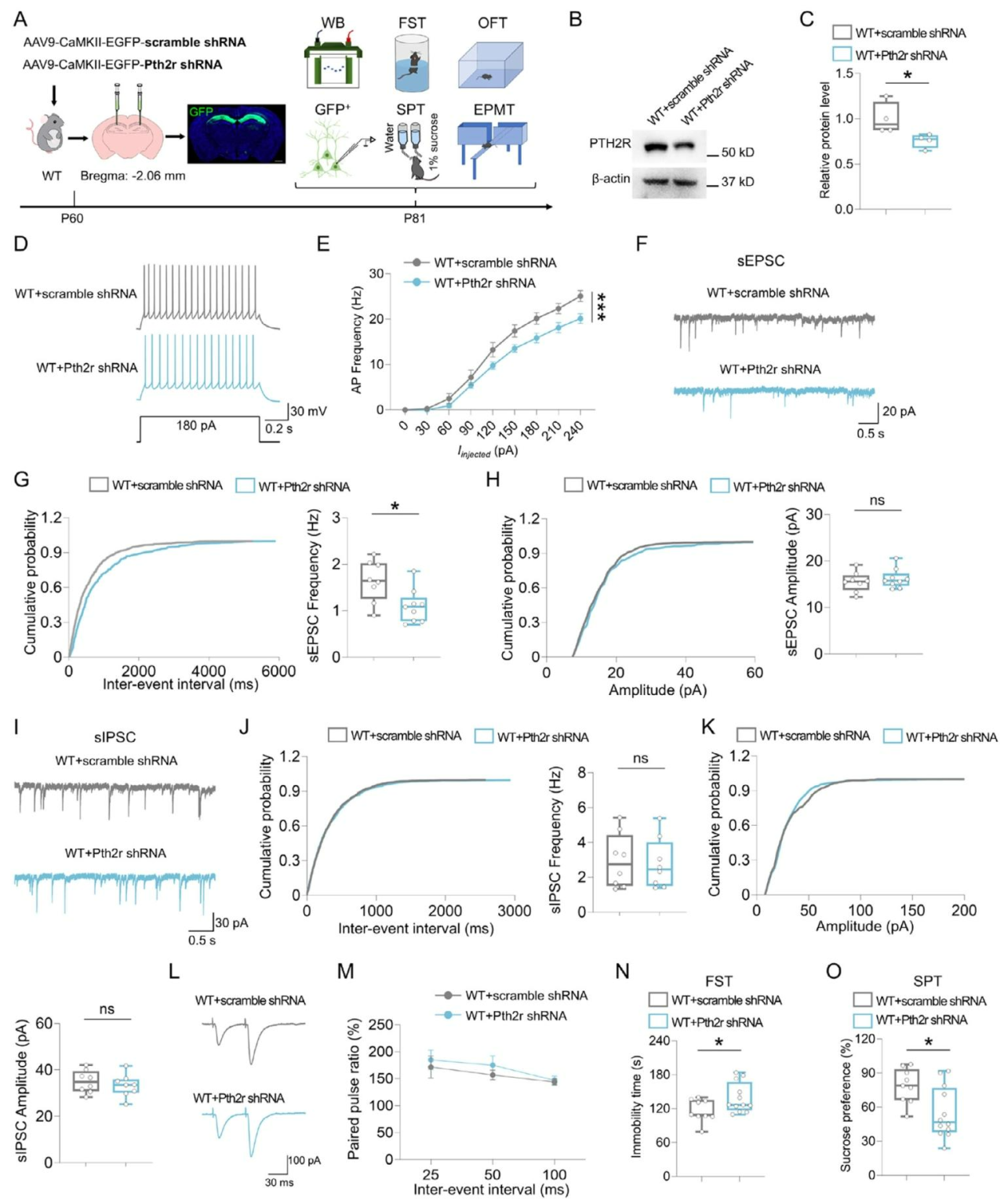
图7 敲低Pth2r降低小鼠神经元兴奋性、损害谷氨酸能神经传递并诱导抑郁样行为
为探究PTH2R是否在Adcy8调控通路中具有必要性,研究向Adcy8CaMKII-CKO小鼠双侧dCA1区注射Cre依赖型过表达EGFP或PTH2R AAV(图8A)。3周后,Westernblot证实Adcy8CaMKII-CKO+PTH2R组小鼠海马PTH2R蛋白水平显著回升(图8B、C),其dCA1区神经元降低的兴奋性得以恢复(图8D、E),且受损的sEPSC也被挽救,而sIPSC及PPR无明显变化(图8F-M)。行为学上,过表达PTH2R显著改善Adcy8CaMKII-CKO小鼠在FST和SPT中的抑郁样表型(图8N、O)。此外,对Adcy8CaMKII-CKO小鼠dCA1连续5天双侧输注PTH2R内源性配体TIP39可显著改善小鼠的抑郁样行为。综上,dCA1 Adcy8通过调控TIP39-PTH2R信号通路,调控神经元兴奋性、谷氨酸能神经传递及小鼠抑郁样行为。
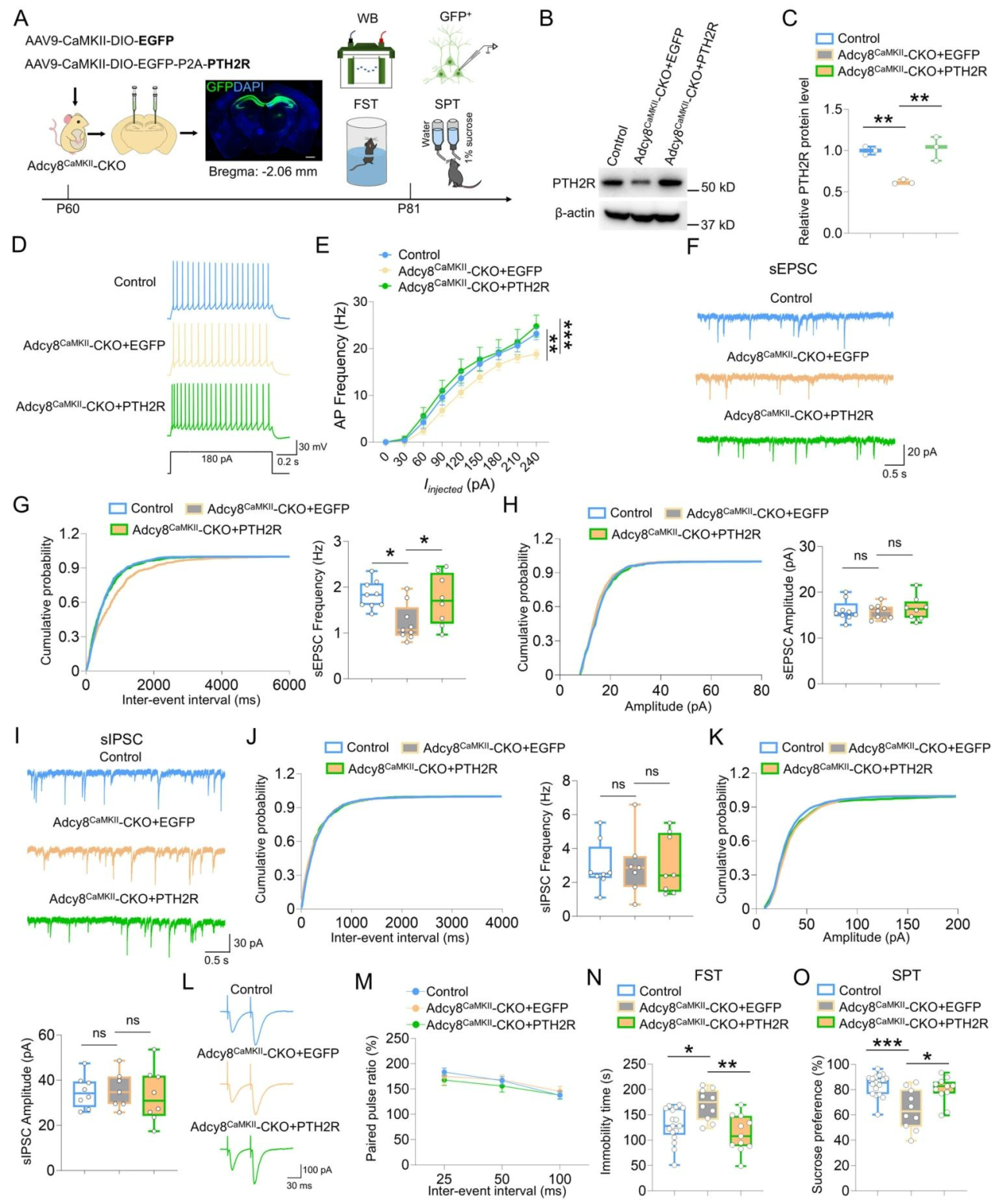
图8 过表达PTH2R可挽救Adcy8CaMKII-CKO小鼠的神经元兴奋性与谷氨酸能神经传递缺陷,并缓解其抑郁样行为。
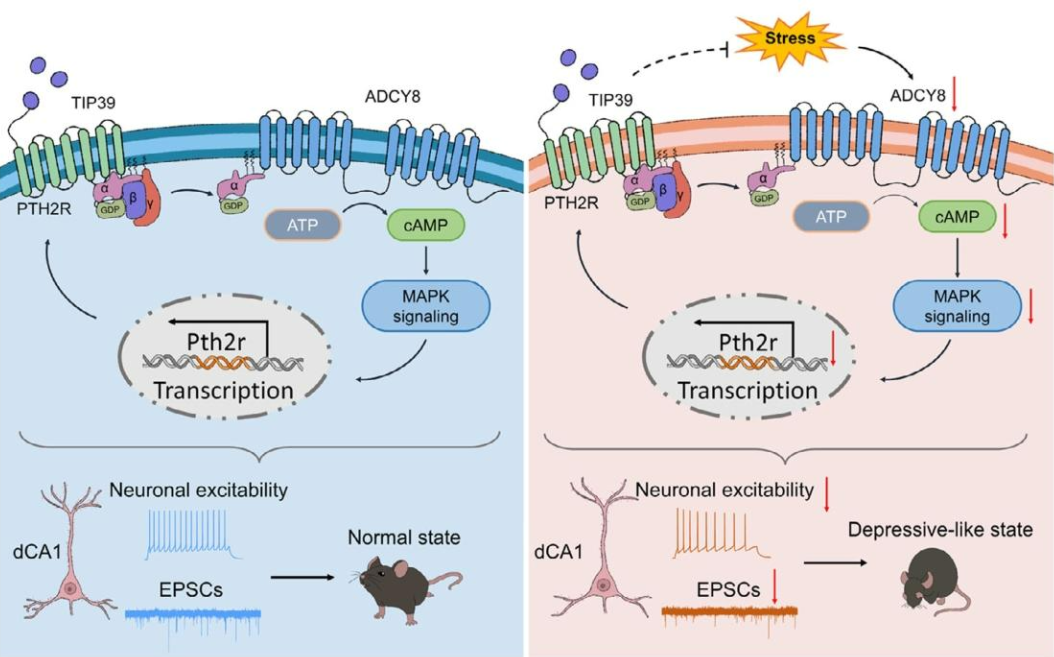
本研究阐明了一条全新的抑郁调控通路:dCA1神经元中Adcy8可通过调控细胞内钙信号、内在兴奋性及谷氨酸能神经传递,发挥抑制小鼠抑郁样行为的作用。相反,慢性应激选择性降低dCA1区兴奋性神经元中Adcy8的表达,其缺失通过抑制MAPK信号下调PTH2R水平,损害神经元兴奋性与谷氨酸能传递,最终诱发抑郁样行为;而干预下游PTH2R或其内源性配体TIP39可逆转相关缺陷,揭示了Adcy8-PTH2R信号轴在应激相关抑郁中的核心作用,为抑郁症提供了潜在治疗靶点。
本文使用的工具病毒布林凯斯均可提供:
同时布林凯斯也可提供各类定制服务请联系小布:18971216876(微信同号)或者咨询所在区域的销售经理获取更多信息。