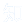脊髓背角(SCDH)神经网络中兴奋与抑制的平衡是维持疼痛处理的关键,神经损伤会破坏这种平衡,抑制性神经传递受抑,导致疼痛信号增强。PKCγ+ 中间神经元位于SCDH lamina II/III,在周围神经损伤和持续炎症后的机械性异常疼痛中起关键作用,正常情况下受抑制性神经元调控,病理状态下抑制解除,引发异常疼痛。小胶质细胞激活后释放神经炎症介质,近年发现其介导的突触修剪通过补体系统参与神经疾病,但其在神经病理性疼痛中对抑制性突触的修剪及其对靶细胞和疼痛处理的影响尚不明确。
2025年6月5日,香港中文大学刘晓东和深圳南山医院疼痛科蒋昌宇副研究员合作在Science Translational Medicine上发表题为“Microglial pruning of glyci-nergic synapses disinhibits spinal PKCγ interneurons to drive pain hypersensiti-vity in mice”相关文章,该研究表明脊髓小胶质细胞通过修剪甘氨酸能突触,导致脊髓PKCγ中间神经元去抑制,进而促进低阈值Aβ纤维的神经传递,引发痛觉过敏。

在神经病理性疼痛SNI模型小鼠中,损伤后出现疼痛超敏(图1A)。损伤后第3天(PID3),同侧SCDH的Iba1阳性小胶质细胞数量显著增加,至PID14仍维持高位;小胶质细胞中溶酶体相关蛋白CD68密度持续升高至少21天。体外实验证实,PID14时SNI小鼠小胶质细胞吞噬能力显著强于假手术组,对突触小体吞噬量更高(图1B-C)。检测突触标志物发现,与假手术组相比,PID14时SNI小鼠SCDH中抑制性突触(GlyT2,标记甘氨酸能神经元;GAD65/67,标记GABA能神经元)前终末密度降低,小胶质细胞密度增加,而兴奋性突触(VGluT1、VGluT2、Homer1,对应谷氨酸能神经元)没有变化(图1D-F)。甘氨酸能神经元胞体密度不变,但相关基因表达升高,抑制性神经元(Pax2+)密度没有差异。
吞噬分析显示,PID14时小胶质细胞溶酶体内抑制性突触前元件显著增多,对甘氨酸能和GABA能突触前元件吞噬量分别增加7.7倍和1.8倍(图1G-H)。SNI小鼠脊髓背角II-III层中小胶质细胞密度与GlyT2免疫反应性呈负相关(图1I-J)。动态刷拭刺激(检测疼痛反应)下,PID14时SNI小鼠中c-Fos+/PKCγ+中间神经元的比例高于假手术组(图1K-L)。PID14时SNI组PKCγ+中间神经元上的甘氨酸能终末数量少于假手术组,而PID7时没有此差异(图1M-N)。在PID7和PID14时,SNI小鼠中激活的小胶质细胞包裹着PKCγ+中间神经元的胞体(图1O-Q)。雌性小鼠验证了相同结果,包括疼痛超敏、小胶质细胞激活、抑制性突触减少及PKCγ+神经元激活。综上,小胶质细胞对甘氨酸能突触前元件的吞噬与PKCγ+中间神经元抑制解除,共同推动神经病理性疼痛发展。
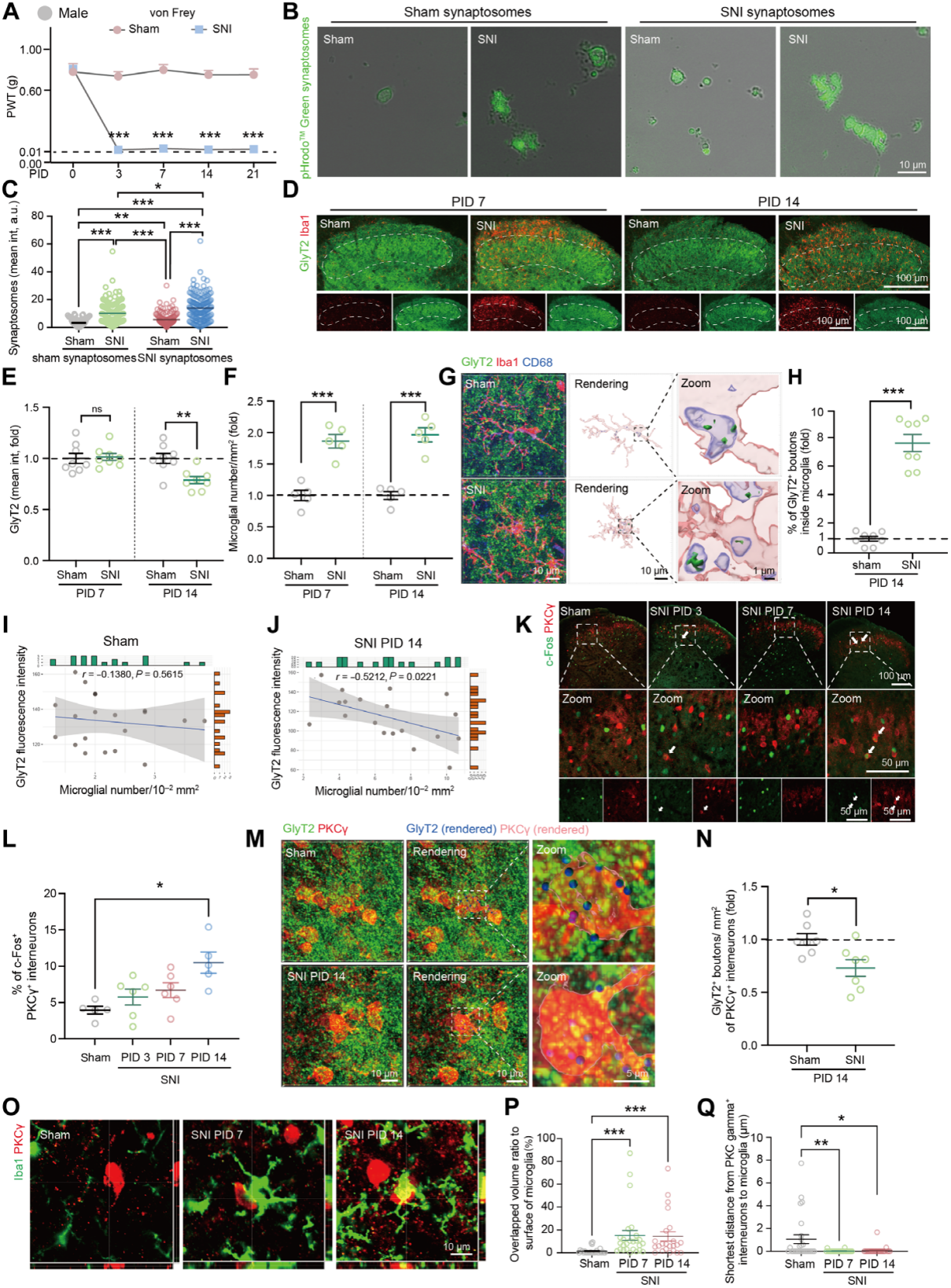
图1 雄性小鼠外周神经损伤后小胶质细胞对抑制性突触前成分的吞噬作用增强及脊髓去抑制现象
为了证实小胶质细胞是否在SNI后对中枢去抑制起关键作用,从假手术/SNI前14天开始,通过灌胃给予集落刺激因子1受体(CSF1R)抑制剂PLX3397,以耗竭SCDH中的髓系细胞(主要是小胶质细胞)(图2A)。结果显示,小胶质细胞耗竭能减轻SNI诱导的机械性超敏反应(图2B-C),逆转了PID14时甘氨酸能突触前终末丢失(图2D-E),消除了SNI诱导的小胶质细胞激活(图2D、F),并减弱了PKCγ+中间神经元上GlyT2阳性斑点减少(图2G-H)。进一步检测发现,体内耗竭小胶质细胞消除了SNI诱导的PKCγ+中间神经元甘氨酸能sIPSC频率降低(图2I-J);与假手术组相比,SNI增加了PKCγ+中间神经元兴奋性,而小胶质细胞耗竭可消除这种过度兴奋性(图2K-N),且对假手术组无影响。
图2 小胶质细胞耗竭可缓解雄性小鼠中SNI诱导的甘氨酸能突触缺失、PKCγ+中间神经元过度兴奋及机械性痛觉过敏
为探究小胶质细胞耗竭能否逆转SNI诱导的异常疼痛,采用光遗传学激活Aβ纤维记录疼痛样行为。使用Thy1.2-cre/ERT2-EYFP小鼠在大直径神经元表达ChR2,将RetroAAV-hSyn-DIO-ChR2-mCherry注射到小鼠的左腰L4-L6脊髓背角,实现胞体和外周皮肤末梢的ChR2表达(图3A),特异性调控Aβ低阈值机械感受器(LTMR)。在PID14时,向同侧后爪的外侧足底表面施加蓝光以刺激Aβ纤维(图3A)。光遗传学激活Aβ纤维在SNI小鼠中诱导的爪退缩率高于假手术组小鼠(图3B),表明存在触觉异常疼痛。小胶质细胞耗竭抑制了SNI小鼠对Aβ纤维激活的反应性(图3B)。同时,Aβ纤维激活在SNI小鼠中诱导更高比例的c-Fos+/PKCγ+中间神经元和I层c-Fos+细胞增加,小胶质细胞耗竭则降低了相关细胞比例和密度(图3C-D)。
为了研究小胶质细胞耗竭是否调节PKCγ+中间神经元对低阈值Aβ初级传入输入的反应性,对PKC gamma-CreERT2:Ai9小鼠脊髓切片中的tdTomato表达神经元进行了全细胞膜片钳记录(图3E-F)。记录表明,PLX3397给药增强了SNI小鼠tdTomato神经元的甘氨酸能eIPSC幅度(图3G-H),但不影响Aβ纤维刺激诱发的eEPSC幅度(图3I-J)。此外,鞘内注射米诺环素抑制脊髓小胶质细胞激活,可减轻疼痛敏感性,逆转GlyT2+突触终末丢失,并抑制相关神经元及LPBN细胞激活。对照实验(脾切除术、鞘内注射生理盐水)排除了操作假象和外周免疫细胞浸润的影响。综述所述,小胶质细胞介导的抑制性突触吞噬功能受阻,一定程度上促进了脊髓抑制回路的结构与功能恢复。
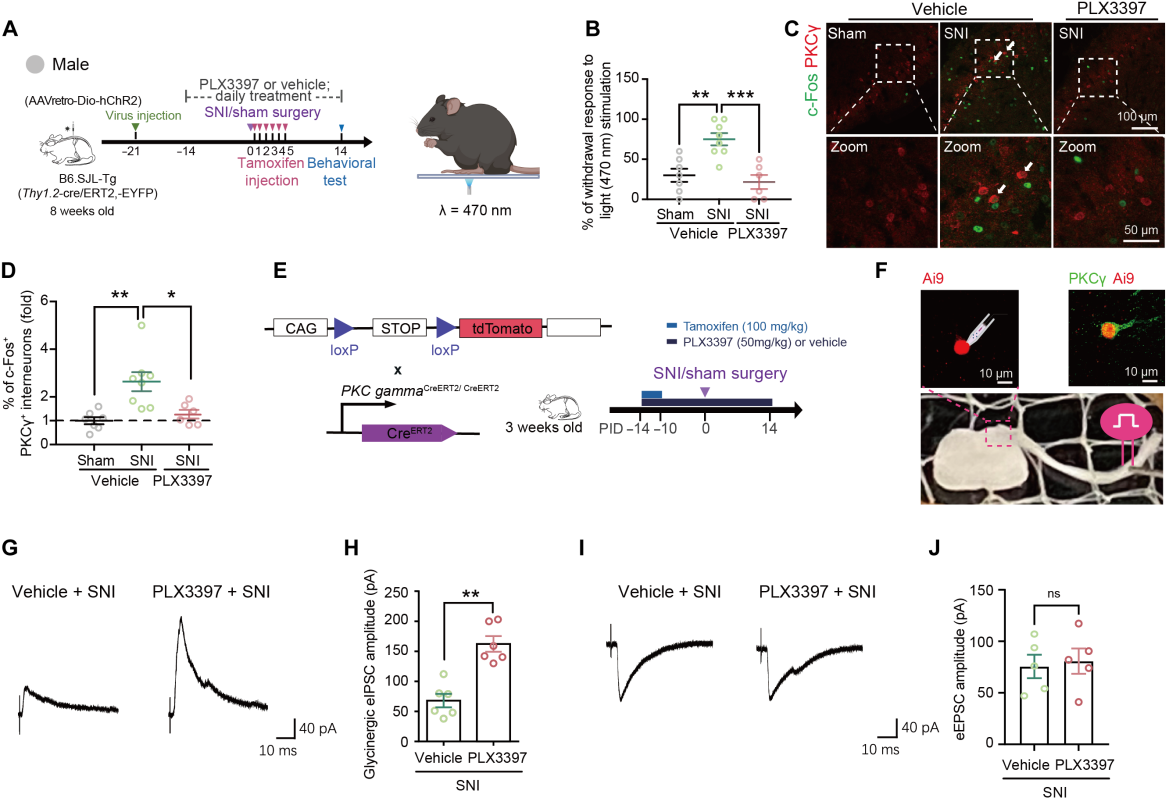
图3 小胶质细胞耗竭抑制了SNI诱导的对Aβ纤维激活的行为和神经元反应增强
补体系统通过标记突触并发出“吞噬我”或“不吞噬我”的信号来影响突触修剪。SNI后,雄性和雌性小鼠SCDH及SCDH分离的小胶质细胞中C1q表达均增加。此外,与假手术组相比,SNI小鼠SCDH中与甘氨酸能突触前小体共定位的C1q免疫反应信号多约6倍(图4A-B),小胶质细胞吞噬的C1q标记的GlyT2+斑点比例更高(图4C-D)。用C1q中和抗体阻断C1q(图4E),在SNI模型损伤后第14天,该抗体缓解部分机械性超敏反应(图4F-G);逆转甘氨酸能突触前终末丢失(图4H-I),恢复PKCγ+中间神经元相关突触前小体密度(图4J-K)和sIPSCs频率与幅度(图4L-M);此外,抗C1q抗体降低了SNI小鼠中c-Fos+/PKCγ+中间神经元的比例(图4N-O)。
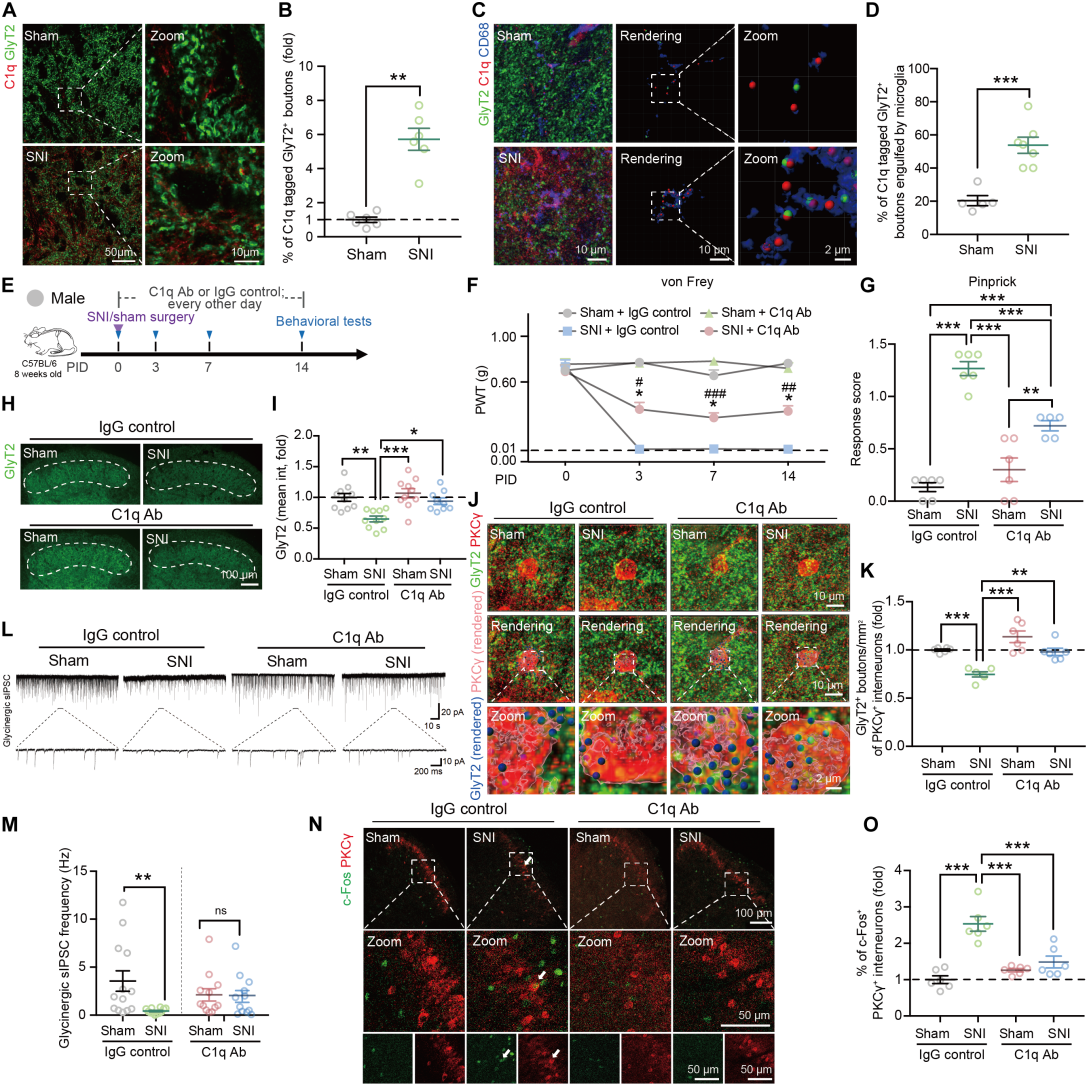
图4 SNI诱导的甘氨酸能突触修剪与机械性痛觉过敏均需依赖C1q信号通路
利用单细胞RNA测序平台,对SNI或假手术小鼠同侧SCDH中经FACS分选的髓系细胞进行转录组分析。在PID3、7、14,取L4-L6水平同侧SCDH组织(图5A),并使用单核细胞/巨噬细胞表面标志物Cd11b进行荧光激活细胞分选Cd11b+细胞测序。将小胶质细胞分为16个簇(MG0-15),排除3个次要簇(图5B)。通过鉴定各簇差异表达基因(DEGs)发现(图5D),MG1和MG3在PID7出现并持续至PID14(图5C)。MG1稳态及增殖基因低表达,H2-D1和B2m高表达(图5D);MG3代谢等稳态基因低表达,Apoe、Lyz2和Cd63高表达(图5D)。对SNI-PID7和14样本中的MG1、MG3进行GO富集分析(图5E-G),MG3的DEGs在突触修剪通路显著富集(图5G),且富集吞噬相关基因(Axl)和肝X受体(LXR)靶基因(如Apoe、Abca1和Abcg1)(图5F和H)。经qRT-PCR和免疫荧光染色验证,SNI后雌雄小鼠SCDH中Apoe表达在PID7和14升高,APOE+小胶质细胞比例增加(图5I-L),且几乎所有APOE+小胶质细胞与LXR共定位(图5M-P),其甘氨酸能成分多于APOE−细胞(图5Q-R)。此外,PID14时无APOE表达的小胶质细胞仍呈激活态,表明APOE是非激活通用标志物,且MG3转录谱与疾病相关小胶质细胞(DAM)相似。
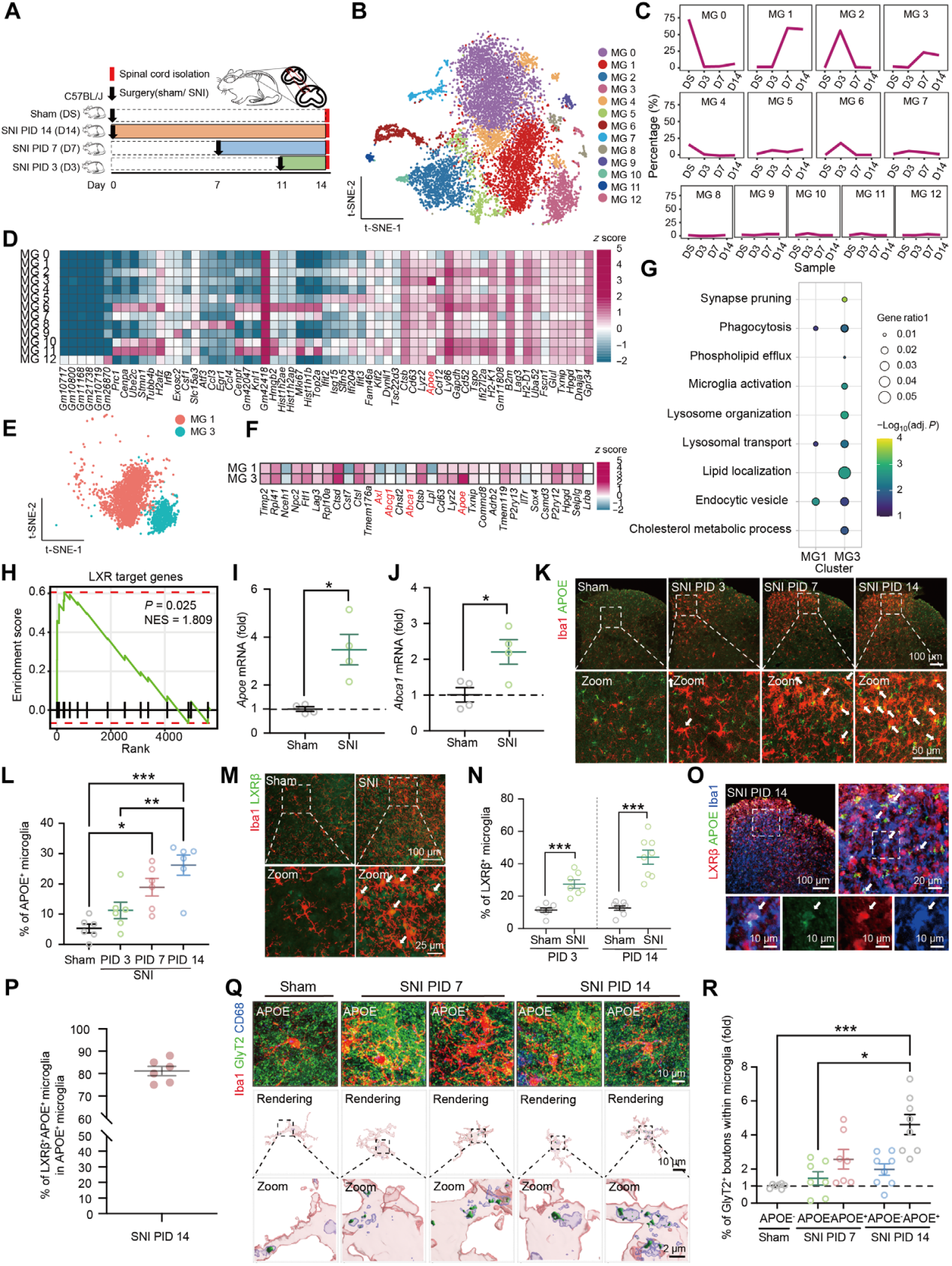
图5 表达LXR/APOE的小胶质细胞在坐骨神经损伤后被上调并吞噬甘氨酸能突触
已知Apoe是LXR下游转录靶点。为了探究LXR-APOE轴是否参与小胶质细胞介导的神经病理性疼痛中SCDH抑制性突触丢失,在永生化小胶质细胞(IMG)系中,LXR激动剂GW3965增强Apoe、Abca1和Abcg1表达,提升细胞吞噬活性。这些基因在单细胞RNA测序数据中属于APOE主导的小胶质细胞亚群的上调基因。构建慢病毒载体(LV-hCD68-shRNAmir-Apoe)实现小胶质细胞特异性Apoe敲低。体外实验中,Apoe敲低消除GW3965诱导的吞噬活性;小鼠实验显示,注射靶向Apoe的慢病毒可下调SCDH小胶质细胞Apoe表达(图6A),减少APOE+小胶质细胞数量,缓解SNI诱导的SCDH甘氨酸能突触前元件丢失、与PKCγ+中间神经元的锚定减少(图6B-E),以及c-Fos+/PKCγ+细胞增加(图6F-G)。
为确定小胶质细胞APOE是否参与维持痛觉过敏,不同时间敲低Apoe发现,SNI前或早期敲低均可减轻痛觉过敏,且雌性小鼠结果类似。Apoe全身敲除小鼠实验表明,Apoe缺失消除SNI诱导的小胶质细胞突触体吞噬增加,逆转甘氨酸能突触丢失,抑制PKCγ+中间神经元激活。在SCDH小胶质细胞特异性Apoe敲低小鼠(图6H-J)和Apoe缺陷小鼠中评估痛觉样行为。与野生型小鼠或对照同窝小鼠相比,Apoe缺失或小胶质细胞特异性Apoe敲低可减轻SNI诱导的动态触觉异常疼痛、针刺反应和机械性异常疼痛。通过鞘内注射CSF1联合GW3965,增加小鼠SCDH中APOE+小胶质细胞比例,导致小胶质细胞内CD68+溶酶体增多、GlyT2+抑制性突触密度降低,且激活的小胶质细胞溶酶体内GlyT2+残留物增加。
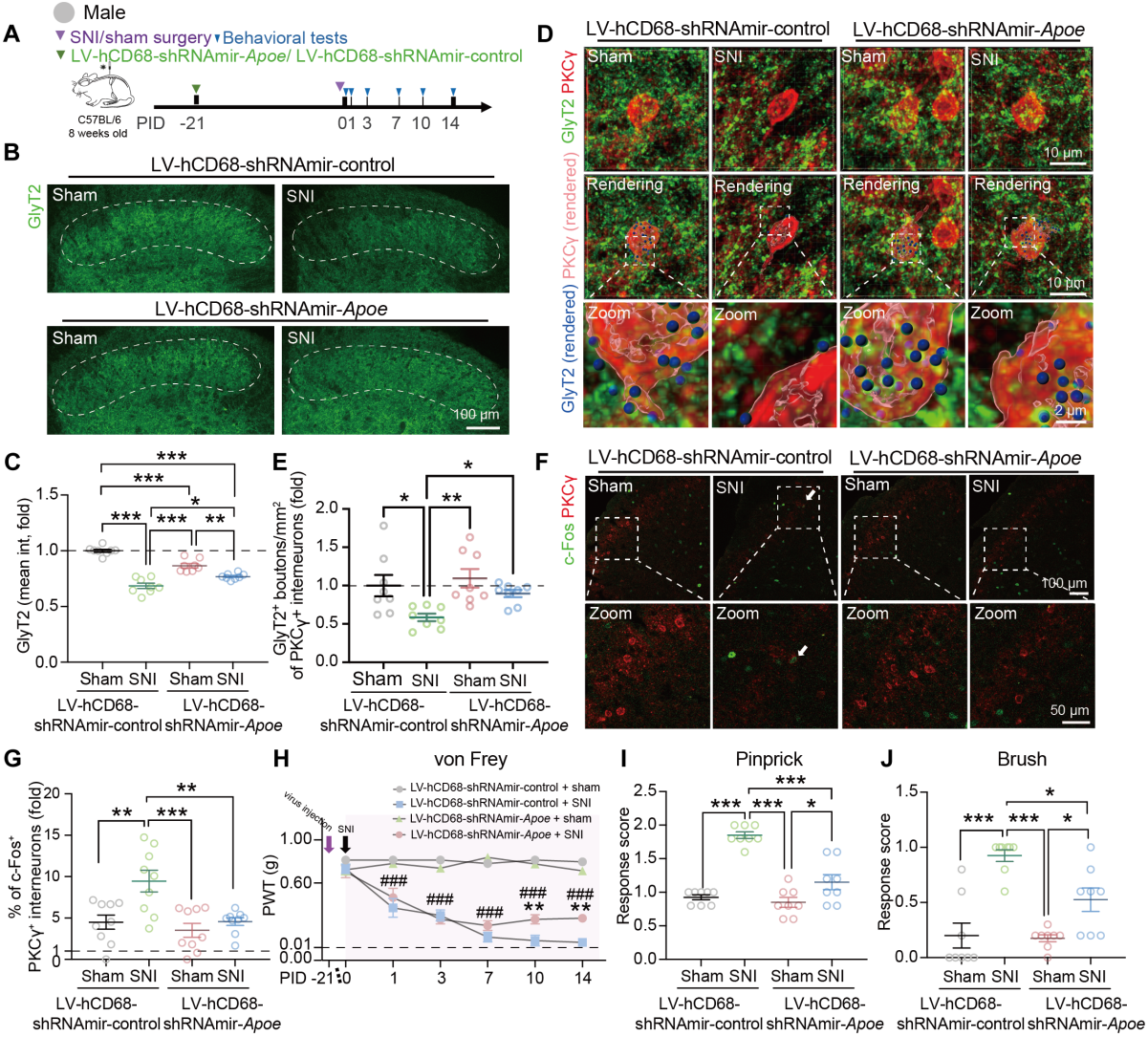
图6 小胶质细胞APOE的缺失抑制SNI诱导的甘氨酸能突触修剪并减轻机械性超敏反应
为探究LXR-APOE轴对小胶质细胞C1q产生的调控作用,用LXR激动剂GW3965和拮抗剂GSK2033处理IMG细胞,发现C1q及Apoe、Abca1、Abcg1表达相应改变。CSF1与GW3965联合处理小鼠后,其SCDH中C1q表达增强,对照处理组无此变化,且GSK2033抑制SNI诱导的SCDH小胶质细胞内C1q密度增加(图7A-C)。基于SNI小鼠中C1q表达与小胶质细胞APOE密度正相关(图7D-E),进一步验证APOE对C1q的调控。结果显示,IMG细胞敲低Apoe后,GW3965诱导的C1q相关基因表达下降(图7F-H);Apoe−/−小鼠原代小胶质细胞的C1q相关基因表达改变。小胶质细胞特异性Apoe敲低小鼠及Apoe−/−小鼠中,SNI诱导的脊髓小胶质细胞C1q产生均减少(图7I-N)。
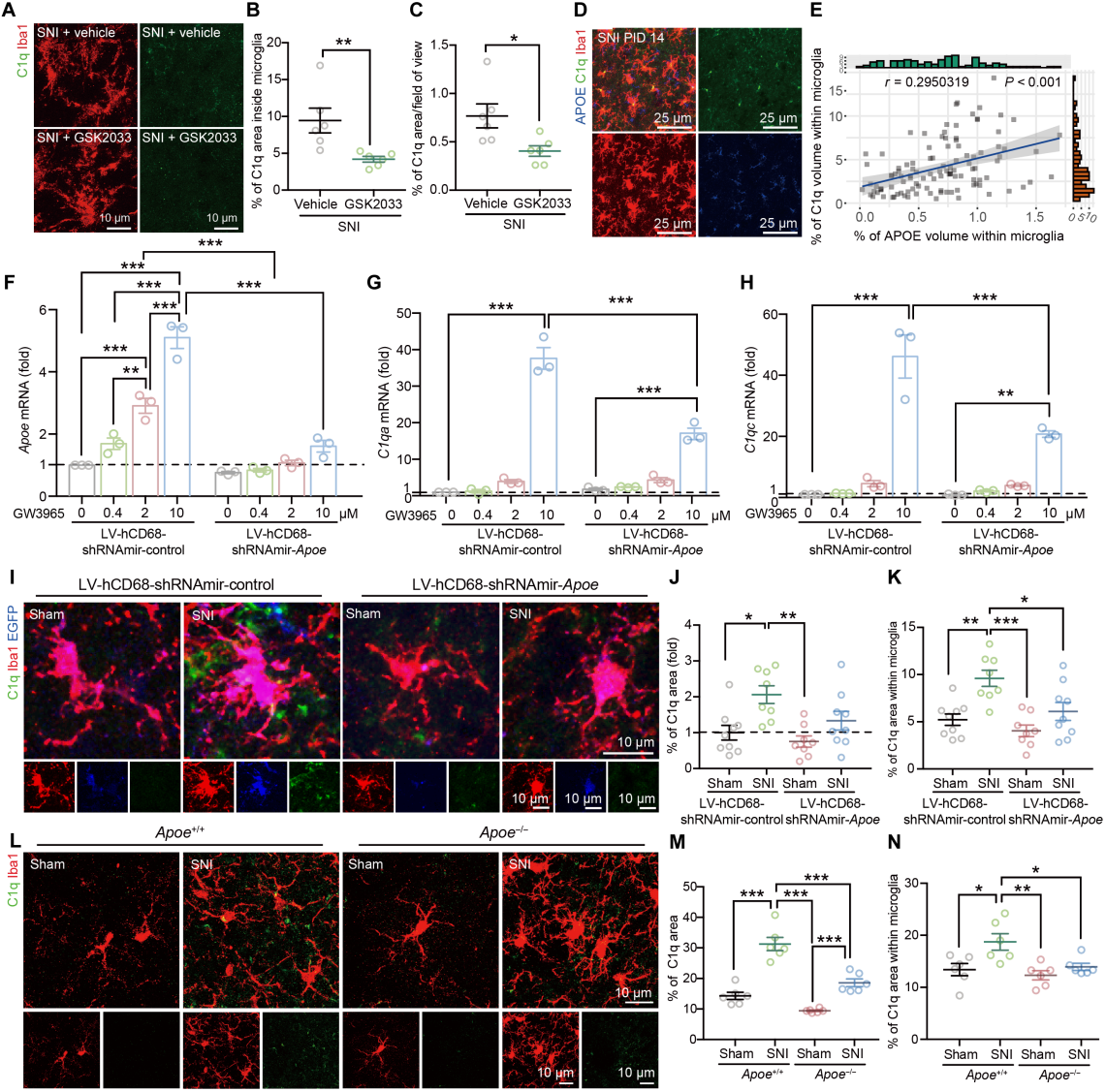
图7 小胶质细胞内的LXR-APOE轴调控补体激活
本文在小鼠SNI模型中发现,脊髓小胶质细胞通过修剪甘氨酸能突触,导致脊髓PKCγ中间神经元去抑制,进而促进低阈值Aβ纤维的神经传递,引发痛觉过敏;其中,小胶质细胞高表达的Apoe和补体C1q是关键介质,敲除Apoe、小胶质细胞特异性敲低Apoe或使用抗C1q抗体可逆转上述过程,减轻痛觉过敏。这一发现揭示了小胶质细胞介导的脊髓抑制性突触修剪在神经性疼痛发展中的作用机制,为通过恢复抑制性门控控制来治疗痛觉过敏提供了潜在的治疗靶点和策略。
本文使用的工具病毒布林凯斯均可提供:

同时布林凯斯也可提供各类定制服务请联系小布:18971216876(微信同号)或者咨询所在区域的销售经理获取更多信息。