动物造模是生命科学和医学研究的核心手段,通过模拟人类生理与疾病,规避人体实验限制,为解析机制、研发药物提供可控平台。动物造模是连接基础研究与临床转化的关键桥梁,意义重大。
传统动物模型主要包括转基因动物、化学诱导、手术造模、自发性疾病模型(自然突变/育种),但存在周期长、成本高、操作复杂等问题。重组腺相关病毒(AAV)作为基因治疗中的递送载体,在动物模型构建中也展现出明显优势。
周期快且操作简便
可直接通过体内注射(如脑部、静脉)方式高效递送基因,无需复杂胚胎操作,周期快且效率高。
表达持久稳定
病毒基因组以附加体形式长期稳定存在于细胞中,可实现长周期(数月至数年)的基因表达,适于慢性疾病研究。
靶向精准:
不同血清型具有不同组织亲嗜性,可进一步通过特异性启动子或衣壳改造,实现细胞级别精准靶向。
策略灵活多样:
适用于功能获得(过表达)、功能缺失(RNAi)及基因编辑(CRISPR)等多种建模策略,可实现时空特异的基因调控。
应用范围广:
适用于小鼠、大鼠及非人灵长类等多种实验动物,便于在生理结构更接近人类的模型中开展研究。
技术成熟:
生产工艺及使用方案相对成熟稳定,商业化程度高,成本低,易于实验室开展和应用。
AAV因其周期短,操作简单,效率高、策略灵活等优势,已成为构建疾病动物模型,特别是神经、代谢等疾病模型的常用工具。
PD是一种常见于中老年人的中枢神经系统退行性疾病,该病主要影响中老年人的神经系统,导致一系列运动和非运动症状。主要病理特征为黑质致密部多巴胺能神经元进行性变性死亡,且残存神经元内出现以α-突触核蛋白(α-syn)异常聚集为主要成分的路易小体和路易神经突。在疾病模型构建中,常用携带α-syn基因(表达野生型或点突变型如A53T)的AAV等作为工具病毒,通过在特定脑区过表达α-syn,模拟其异常聚集及致病过程,从而建立帕金森病相关动物模型。
案例文献
代谢型谷氨酸受体5通过抑制α-突触核蛋白诱导的小胶质细胞炎症来保护帕金森病中的神经毒性[1]
实验动物:Sprague-Dawley大鼠
病毒名称:AAV/9-α-syn
注射方案:SN脑立体定位注射,3μl;8.17 x 1013vg/mL ,表达8周
实验结果:为明确代谢型谷氨酸受体5(mGluR5)在α-syn诱导小胶质细胞活化中的体内功能及PD模型的炎症反应,研究将AAV-α-syn或对照AAV-GFP注入大鼠右侧黑质,不同时间点检测其行为及病理分析。结果显示α-syn过表达组大鼠从病毒注射8周起出现明显行为障碍并持续至16周,黑质多巴胺神经元也逐渐减少;且α-syn升高、mGluR5降低,小胶质细胞中炎症因子(iNOS、IL-1β、TNF-α)在10天升高、4周达峰、8周后消失。这表明PD病理早期存在反应性小胶质细胞的局部炎症,与既往研究一致。
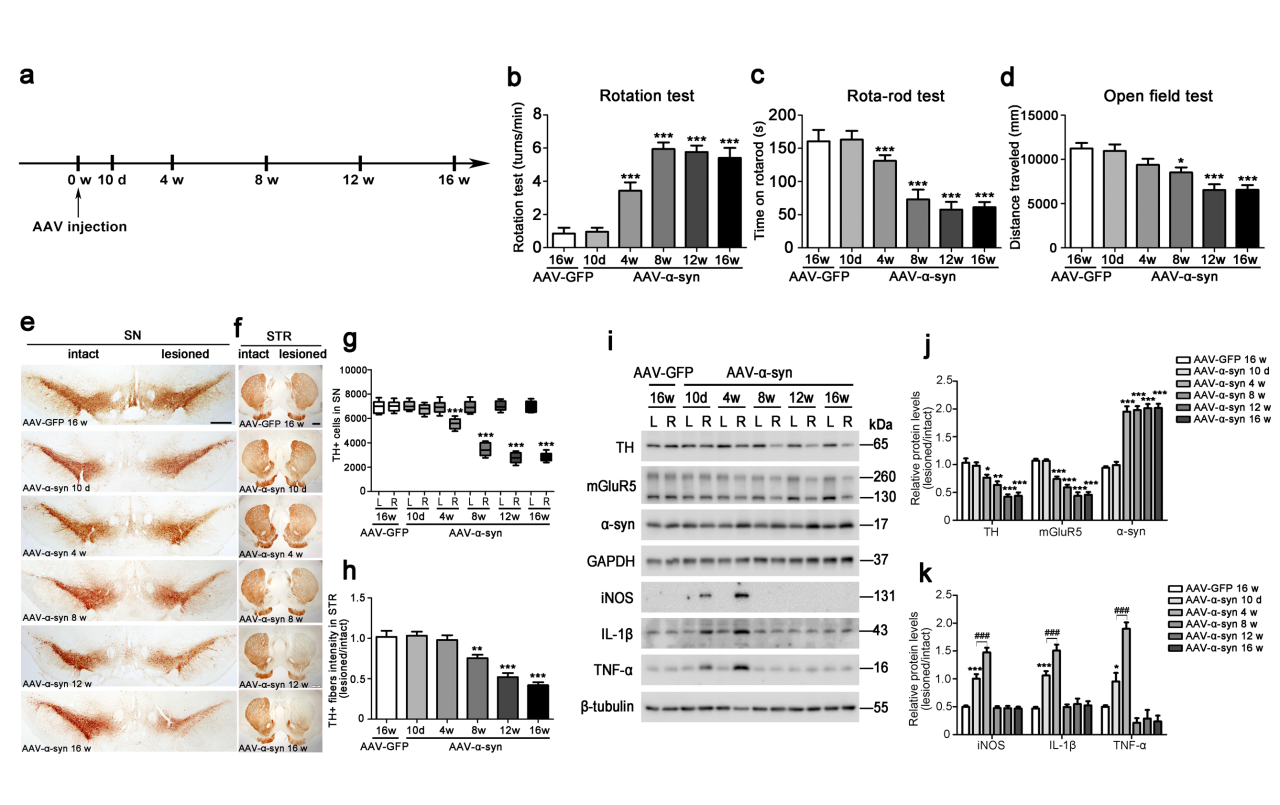
图1. AAV-α-syn诱导大鼠帕金森病模型中进行性神经退行性变的炎症反应
在黑质区通过AAV1/2病毒载体诱导A53T-α-突触核蛋白过表达,可导致黑质纹状体系统变性,伴随路易体样病理改变和运动功能障碍:一种新型帕金森病小鼠模型[2]
实验动物:雄性野生型小鼠
病毒名称:AAV1/2-A53T-α-syn
注射方案:SN脑立体定位注射,1.5μl;5.16 x 1012vg/mL,表达10周
实验结果:以注射AAV1/2-EV的小鼠为对照,注射10周后进行定量分析,结果显示AAV1/2-A53T-α-syn组小鼠同侧黑质中TH +多巴胺能神经元和总神经元数量均显著少于对照组,且同侧纹状体TH + 纤维光密度、突触前DAT水平较对照组分别降低20%和29%。这表明 AAV1/2-A53T-α-syn注射后黑质-纹状体通路发生神经退行性变。
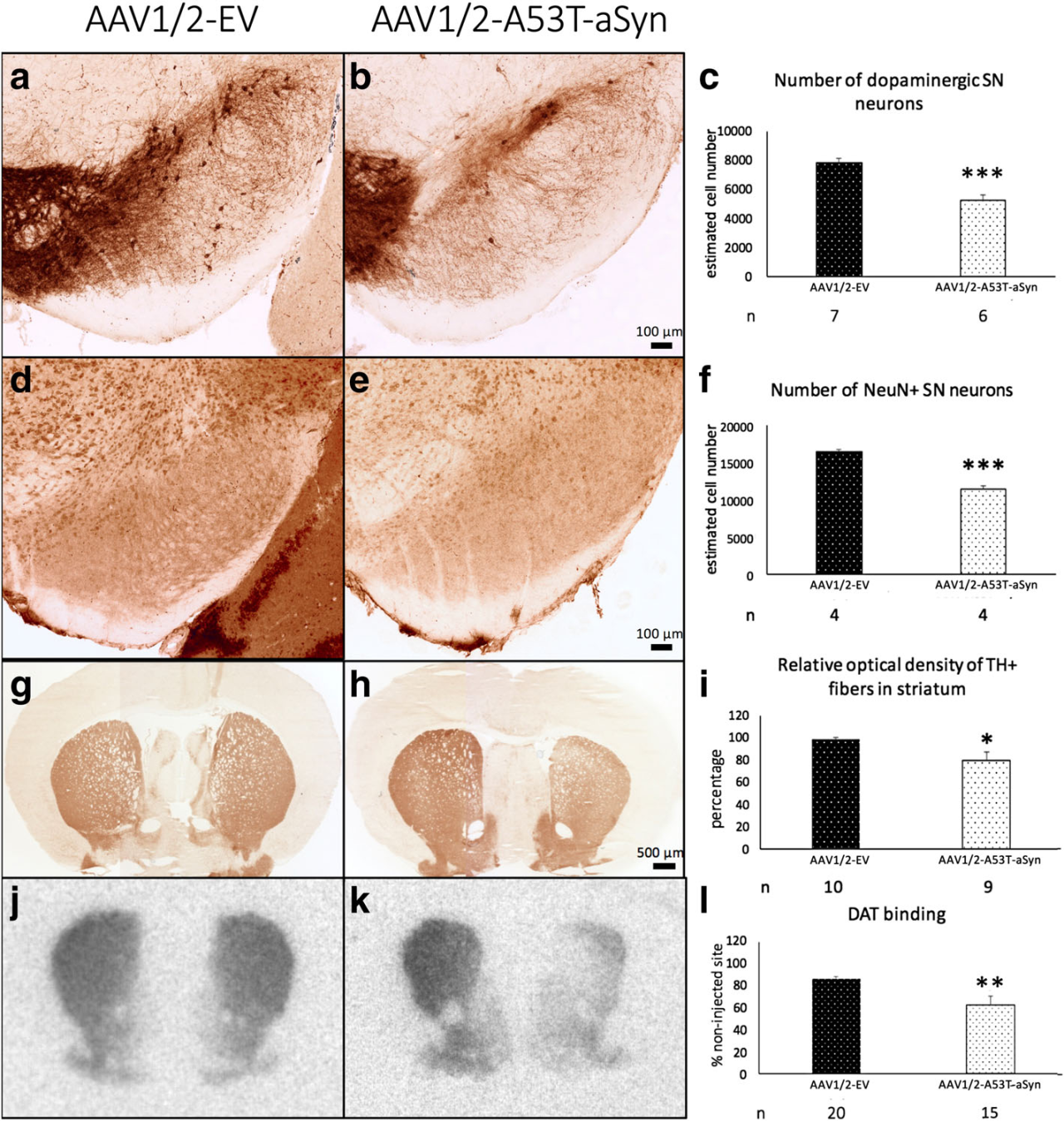
图2 在黑质中注射AAV1/2-A53T-α-Syn会导致黑质纹状体通路发生神经退行性变
AS是一种脂质代谢障碍有关的全身性疾病。它的特点是血液中的脂质进入动脉管壁像粥样斑块沉积,造成动脉狭窄、动脉变硬。在疾病模型构建中,常用携带前蛋白转化酶枯草溶菌素 9(PCSK9)基因(如野生型或突变型)的AAV作为工具病毒,通过静脉注射或局部血管注射,使PCSK9在肝脏或血管组织过表达,以促进低密度脂蛋白胆固醇(LDL-C)代谢异常、加速动脉壁脂质沉积,从而构建动脉粥样硬化动物模型,常与高脂饮食联合使用以增强造模效果。
案例文献
无需生殖系遗传工程改造在小鼠和大鼠中诱导动脉粥样硬化[3]
实验动物:C57小鼠
病毒名称:rAAV8-D377Y-mPCSK9
注射方案:尾静脉注射,2.0 x 1010vg、1.0 x 1011vg或 5.0 x 1011vg,表达12周
喂养方式:西式饮食——21%脂肪和0.21%胆固醇;Paigen饮食——含16%脂肪、1.25%胆固醇和0.5%胆酸钠
实验结果:rAAV8-D377Y-mPCSK9注射12周后处死小鼠,对动脉粥样硬化程度进行定量分析。结果显示对照组小鼠无论采用何种饮食,均无病变或仅有极轻微病变;而所有注射rAAV8的小鼠均以剂量依赖性方式出现动脉粥样硬化。对主动脉根部病变的组织学分析显示,病变处已形成包含泡沫细胞、平滑肌细胞及纤维组织的晚期动脉粥样硬化斑块,除1只注射高剂量 rAAV8-D377Y-mPCSK9的小鼠和1只低密度脂蛋白受体敲除(Ldlr−/−)小鼠外,其余所有小鼠均出现坏死核心形成。
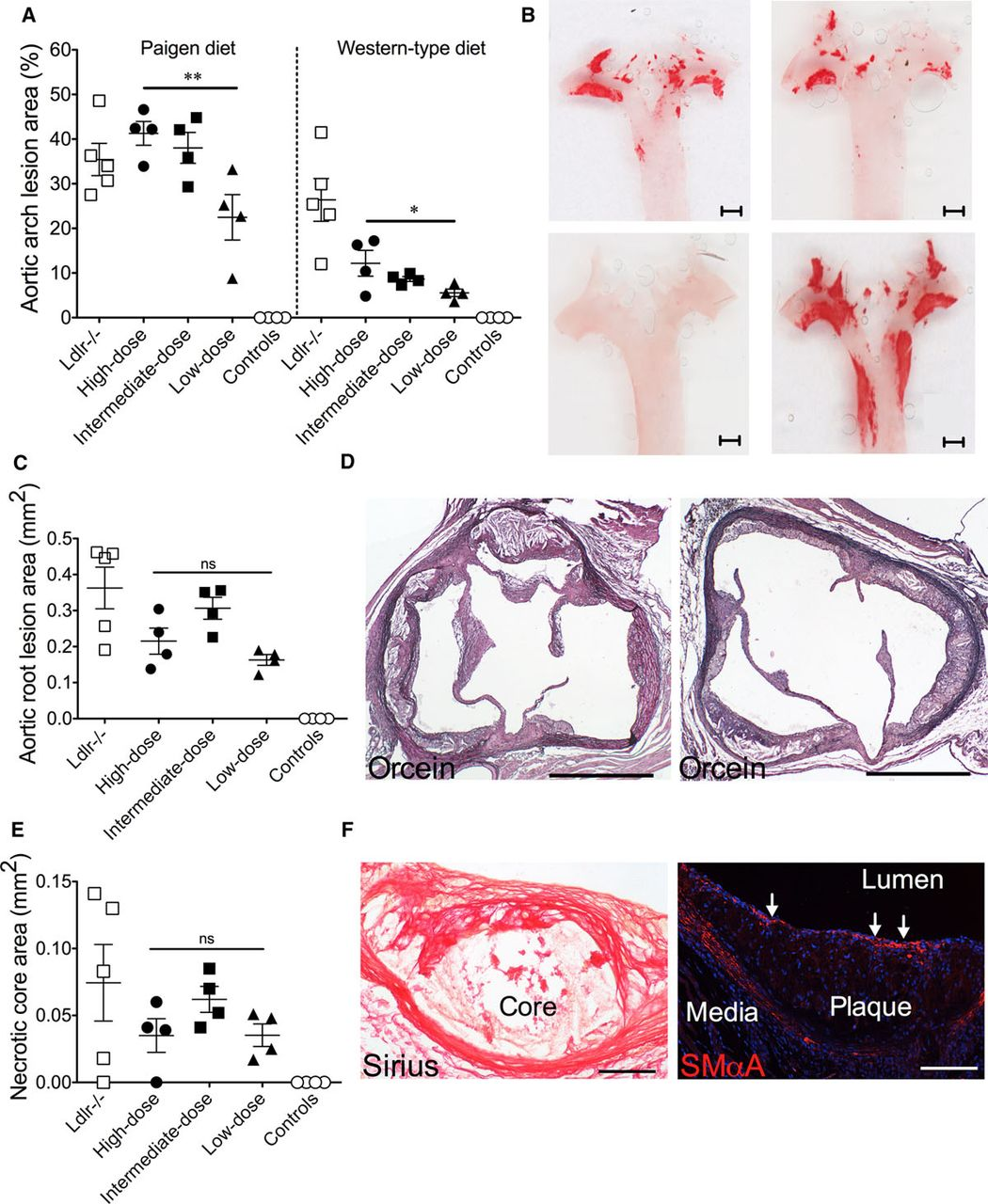
图3 rAAV8-D377Y-mPCSK9诱导的动脉粥样硬化
通过CYSLTR2和P2RY6感知神经酰胺以加剧动脉粥样硬化[4]
实验动物:C57BL/6J品系小鼠
病毒名称:rAAV8-D377Y-hPCSK9
注射方案:尾静脉注射,2.0 x 1011vg,表达12周
喂养方式:HFD——常规酪蛋白配方,含1.25%胆固醇和0.5%胆酸钠
实验结果:为探究CYSLTR2和P2RY6是否介导神经酰胺对动脉粥样硬化的加重作用,实验以8周龄WT、Cysltr2-/-、P2ry6-/-及双敲除小鼠为对象,对其注射 AAV8-hPCSK9-D377Y并饲喂高脂饲料12周,另设WT小鼠注射AAV-Control + 普通饲料为空白组,10周龄起对“AAV-PCSK9 +高脂”小鼠静脉注射 C16:0神经酰胺(5mg/kg/48h)。结果显示,C16:0神经酰胺不影响血脂但使血浆神经酰胺翻倍,显著加剧WT小鼠斑块形成,单敲除小鼠该效应部分减弱,双敲除小鼠斑块、脂质堆积及巨噬浸润进一步减少。CYSLTR2和P2RY6受体可协同介导神经酰胺对动脉粥样硬化的加重作用。
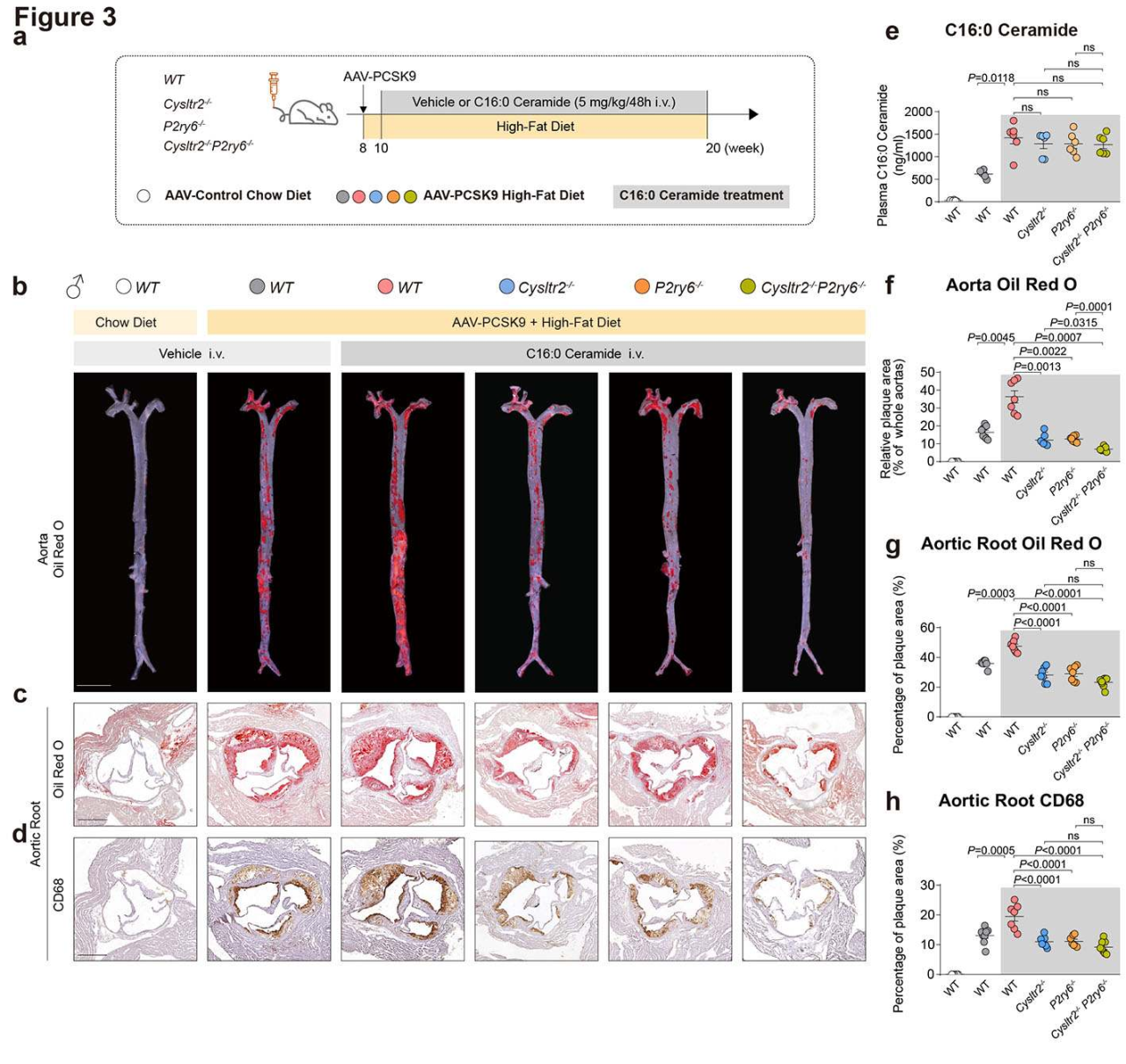
图4 敲除CYSLTR2/P2RY6可减轻神经酰胺加重的动脉粥样硬化
AD是常见的老年神经退行性疾病,以进行性认知衰退和神经损伤为核心,症状呈阶段性加重。神经病理学上,AD大脑内β-淀粉样蛋白(Amyloid β, Aβ)沉积形成老年斑、过度磷酸化Tau蛋白聚集形成神经原纤维缠结(Neurofibrillary tangles, NFT),以及神经元丢失与突触损伤。在AD研究中,常用腺相关病毒AAV作为载体,将APP突变基因、Tau突变基因等AD病理相关基因递送至实验动物脑内(如海马、皮层),诱导β-淀粉样蛋白沉积、Tau聚集及认知障碍,构建贴近AD病理与表型的动物模型,用于机制研究和药物筛选。
案例文献
AAV载体介导的Tau蛋白表达通过细胞周期重入机制导致野生型小鼠锥体神经元变性而不伴随神经原纤维缠结形成[5]
实验动物:野生型FVB/N小鼠
病毒名称:rAAV1/2-hsyn-hTau(P301L)
注射方案:海马区脑立体定位注射,1.0 x 108vg,表达12周
实验结果:向小鼠脑内注射AAV-Tau(P301L),低剂量会致轻度神经退行,海马CA1/2区变薄,神经元丢失越多则hTau越少,因退化神经元蛋白合成减弱。病毒注射1.5周,CA2区先出现病变,3周进展至CA1区,6-12周CA区锥体细胞几乎全丢且延伸至皮层。FJB 染色提示退化神经元,后期小胶质细胞暂态活化,12周消失;星形胶质细胞持续活化显炎症;对照注射AAV-EGFP的小鼠无神经元丢失或小胶质活化。AAV-Tau-P301L会致海马CA区和皮层锥体细胞退化,且小胶质活化仅与Tau介导的神经退行相关,与淀粉样蛋白、病毒颗粒/蛋白反应无关。
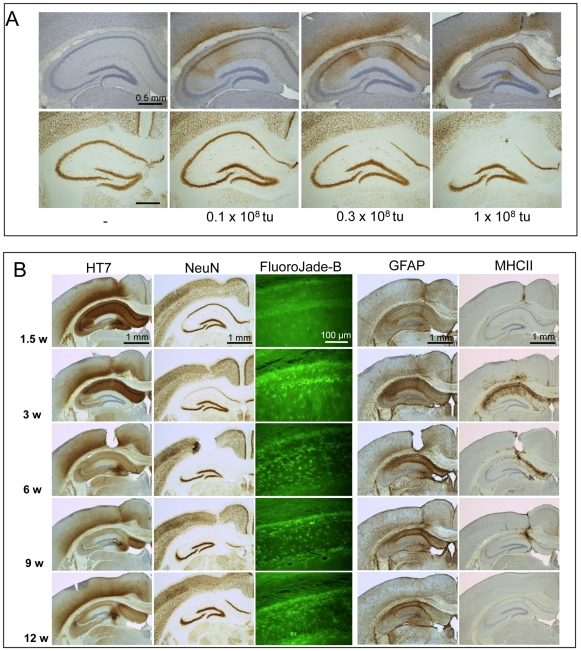
图5 AAV-Tau.P301L介导的神经变性的剂量依赖性和时间线
通过海马区过表达人源tau蛋白构建伴有阿尔茨海默病样病理的非人灵长类动物模型[6]
动物模型:成年恒河猴(7-15岁)
病毒名称:rAAV9-hTau(WT)
注射方案:双侧海马区脑立体定位注射,1010~11 GC ,表达6-12周/50周
实验结果:为构建Tau蛋白病诱导的、含AD样病理的非人灵长类(NHP)模型,选用成年恒河猴(7-15岁),通过立体定向注射过表达hTau蛋白的AAV载体,使用免疫显色及PET/MRI成像等多种检测方法,结果显示AAV介导的基因转导效率约为75%,且在6至50周内保持稳定,表明AAV能够有效诱导tau蛋白的广泛且持久的表达。
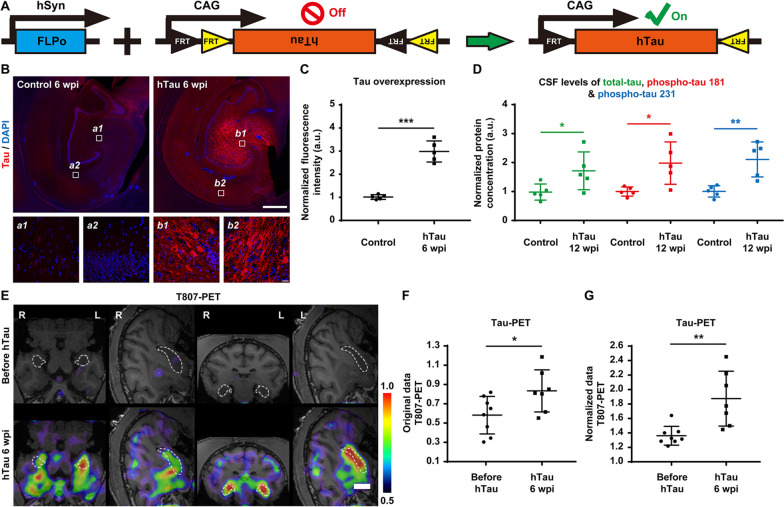
图6脑立体定位将AAV注射至双侧海马体获得tau持续高表达的NHP模型
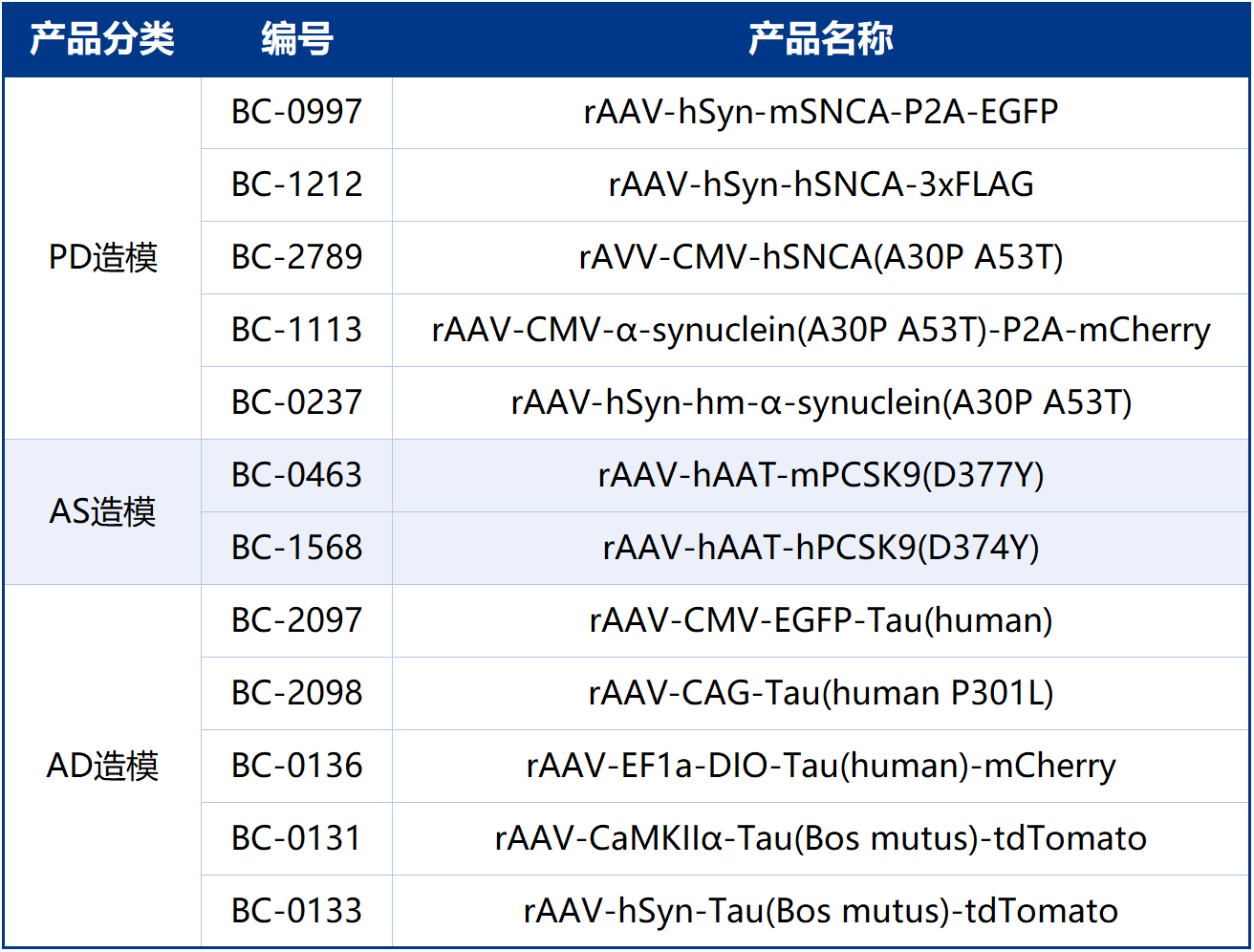
相关AAV造模工具病毒布林凯斯均可提供:
同时布林凯斯也可提供更多定制服务,请联系小布:18971216876(微信同号)或者咨询所在区域的销售经理获取更多信息。
参考文献
[1] Zhang YN, Fan JK, Gu L, Yang HM, Zhan SQ, Zhang H. Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson's disease. J Neuroinflammation. 2021;18(1):23.
[2] Ip CW, Klaus LC, Karikari AA, et al. AAV1/2-induced overexpression of A53T-α-synuclein in the substantia nigra results in degeneration of the nigrostriatal system with Lewy-like pathology and motor impairment: a new mouse model for Parkinson's disease. Acta Neuropathol Commun. 2017;5(1):11.
[3] Bjørklund MM, Hollensen AK, Hagensen MK, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114(11):1684-1689.
[4]Zhang S, Lin H, Wang J, et al. Sensing ceramides by CYSLTR2 and P2RY6 to aggravate atherosclerosis. Nature. 2025;641(8062):476-485.
[5] Jaworski T, Dewachter I, Lechat B, et al. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS One. 2009;4(10):e7280.
[6] Jiang Z, Wang J, Qin Y, et al. A nonhuman primate model with Alzheimer's disease-like pathology induced by hippocampal overexpression of human tau. Alzheimers Res Ther. 2024;16(1):22.



地址:-
地址:-
