在基因治疗领域,腺相关病毒(AAV)载体的质量直接决定疗法安全性与有效性,而关键质量属性(CQA)是质控体系的核心框架。AAV 载体的 CQA 需系统覆盖鉴别、含量与效价、纯度、杂质、安全性五大维度。这些因素会影响转导效率、生物分布和免疫原性,因此评估这些因素对于治疗成功至关重要。对这些属性进行常规表征可以为产品一致性和安全性提供宝贵的见解。 本文将结合检测分析技术、质控标准及适用场景,深度拆解这五大维度,全面呈现 AAV 质控的核心目标与技术要求。
鉴别是 AAV 质控的 “第一道关卡”,核心目标是确认载体 “真实性”—— 确保生产的 AAV 与设计目标一致,无血清型错配、序列突变或交叉污染,避免 “身份错误” 导致的靶向失效或免疫原性升高。根据ICH Q6B 指南要求,AAV 鉴别需从 “蛋白层面” 与 “核酸层面” 双维度验证,具体标准与检测方法如下:
AAV 衣壳由 VP1、VP2、VP3 按 1:1:10 比例组成,不同血清型(如 AAV2、AAV5、AAV8、AAV9)的衣壳蛋白分子量与氨基酸序列存在差异,是蛋白层面鉴别的核心依据。
常用检测方法:SDS-PAGE、CE-SDS
SDS-PAGE 电泳联合银染技术:通过蛋白变性后分子量差异分离 VP1、VP2、VP3,经银染显色后可观察到三条清晰条带,条带对应的分子量需与目标血清型理论值一致(如 AAV9 的 VP1 约 87 kDa、VP2 约 73kDa、VP3 约62 kDa)。该方法操作简便,但分辨率低,难以精准区分近缘血清型间结构蛋白的微小分子量差异,适用于血清型初步筛查而非精准鉴别。
CE-SDS(毛细管电泳 - SDS):以毛细管为分离通道,通过高压电场实现蛋白分离,结合紫外 / 荧光检测可精准量化 VP1/VP2/VP3 比例(需符合 1:1:10 理论范围,偏差≤10%),分辨率远高于 SDS-PAGE,能有效鉴别 AAV2 与 AAV3 等近缘血清型。目前为 FDA 推荐 “首选方法”。
质控标准:
需排除 “杂衣壳” 污染(如生产中混入其他血清型衣壳);若为靶向改造衣壳(如 AAV-PHP.B),需通过肽图分析(HPLC-MS)确认氨基酸序列与设计一致,避免基因突变导致功能异常。
AAV 载体功能依赖基因组 “特征序列”—— 包括维持基因组稳定性的反向末端重复序列(ITR)、治疗基因序列(GOI序列)、启动子序列等,序列缺失或突变会直接导致载体失效。因此,核酸层面鉴别需重点验证关键序列的完整性与正确性。
常用检测方法:PCR 、基因测序、DNA印迹法(Southern Blot)
PCR(聚合酶链式反应):针对 ITR 或治疗基因特异性片段(GOI序列)设计引物,通过扩增产物的电泳条带大小是否符合预期,判断相应的序列存在性。
基因测序:一代测序(Sanger 测序)可精准验证治疗基因编码区等关键区域碱基序列,确保无点突变、插入或缺失;二代测序(NGS):通过对从大批量AAV病毒颗粒中提取的DNA进行建库和NGS测序,可以一次性获得整个基因组的完整序列,精确发现任何单核苷酸变异(SNV)、插入或缺失(Indel)。三代测序(如 PacBio 单分子实时测序)能覆盖完整基因组(约 4.7 kb),解决一代测序难以跨越的 ITR 二级结构问题(ITR 含发夹结构易致测序中断),尤其适用于双链 AAV、自我互补 AAV(scAAV)等复杂载体验证。
DNA印迹法:利用标记的核酸序列作为“探针”,通过核酸杂交(碱基互补配对原则)来检测DNA片段中是否存在特定的基因序列、分析基因的结构、拷贝数以及是否发生重排、缺失等。
质控标准:
特征序列测序覆盖率需达 100%,碱基准确率≥99.99%,且需排除 “野生型 AAV 序列” 污染(野生型含 rep/cap 基因,可能引发免疫反应)。
AAV 载体 “含量” 反映总量,“效价” 反映功能性载体数量,二者共同决定临床给药剂量 —— 剂量不足致疗效失效,剂量过高可能引发免疫反应(如衣壳蛋白诱导抗体反应)。因此,含量与效价的精准量化是 AAV 质控 “核心指标”,需同时检测 “基因组滴度”“衣壳滴度”“感染滴度” 与 “生物学活性”。
基因组滴度(Genomic Titer,GT)指每毫升样品中含完整基因组的 AAV 颗粒数,以每毫升基因组拷贝数 (GC/mL) 或每毫升载体基因组数 (vg/mL)作为单位。
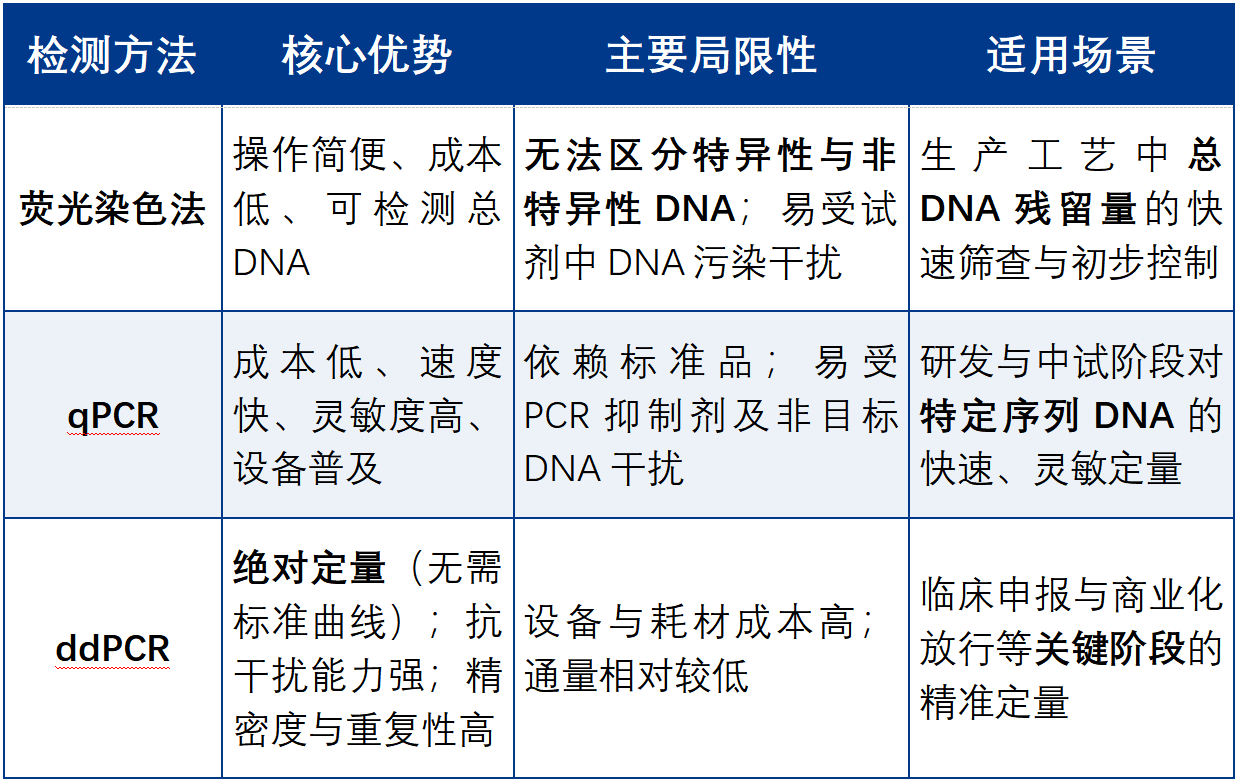
常用检测方法:荧光染色法、qPCR 与 ddPCR
荧光染料法:双链DNA荧光染料与双链DNA特异结合形成复合物,在波长480nm激发下产生超强荧光信号,可用荧光酶标仪在波长520nm处进行检测,在一定的DNA浓度范围内以及在该荧光染料过量的情况下,荧光强度与DNA浓度成正比,根据供试品的荧光强度,计算供试品中的DNA残留量。
qPCR(实时荧光定量 PCR):基于ITR或GOI序列进行荧光定量扩增,通过与标准曲线比较计算滴度。该方法快速、经济,但易受游离质粒或非完整基因组干扰,需在检测前使用DNase I处理样品以降解未包装DNA。。
ddPCR(数字 PCR):将样品稀释至单分子水平,通过微滴化反应实现 “绝对定量”,无需标准品校准,抗干扰能力强,重复性高,已被监管机构推荐作为基因组滴度测定的金标准。
衣壳滴度(Capsid Titer,CT)指每毫升样品中 AAV 衣壳颗粒总数(无论它们是否含有基因组),这对于评估批次一致性和纯化效率至关重要。衣壳滴度以每毫升病毒颗粒数 (vp/mL) 表示。比较衣壳和基因组滴度有助于确定满衣壳与空衣壳的比率,这是载体质量的一个关键指标。
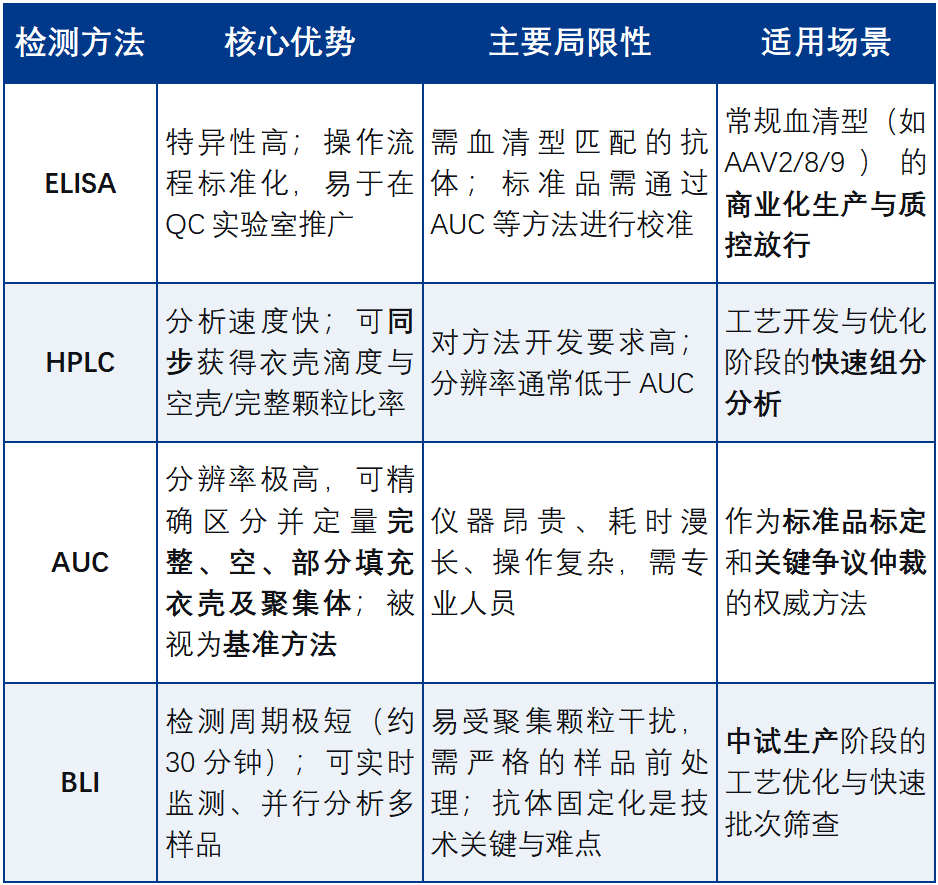
常用检测方法:ELISA 、HPLC、 AUC、BLI
ELISA(酶联免疫吸附试验):基于衣壳特异性抗体进行免疫检测,具备高特异性与标准化优势,但需使用血清型匹配的抗体及可溯源标准品。
HPLC(高效液相色谱):利用衣壳蛋白自身的物理化学性质进行检测,分析速度快,能在一个实验中同时获得衣壳滴度和空壳/完整颗粒比率。
AUC(分析型超速离心):在强大离心力场下,不同沉降系数的颗粒(如完整衣壳、空衣壳、聚集体)会以不同速度沉降。通过光学系统实时监测,得到沉降速度分布图,精确区分并定量完整衣壳、空衣壳、部分填充衣壳和聚集体。
BLI(生物层干涉技术):将抗衣壳抗体固定在传感器表面,通过检测结合后的光学信号变化实时量化衣壳浓度。无需洗涤步骤,检测周期短(约 30 分钟),可同时分析多个样品,但需控制 “聚集颗粒” 干扰(会导致信号异常升高),检测前需用 0.22 μm 滤膜过滤去除大聚集物。
生物学活性是 AAV 载体 “核心功能指标”,需在细胞 / 动物水平验证治疗基因表达产物的预期生物活性(如酶活性、信号通路调控能力),避免因基因序列突变、启动子失效导致 “功能性失效”。
3.1感染滴度(Infectious Titer,IT)指每毫升样品中能成功感染宿主细胞并表达报告基因的 AAV 颗粒数,单位 “TU/mL”(transducing unit/mL)或 “IU/mL”(infectious unit/mL),直接反映载体 “感染能力”—— 感染滴度过低会致细胞转导效率不足,影响疗效。
常用检测方法:TCID50 、流式细胞术、ICA与活细胞分析
TCID50(50% 组织培养感染剂量法):将 AAV 样品梯度稀释后感染敏感细胞,需同时提供辅助病毒,培养 48-72 小时后通过免疫荧光检测报告基因(如 GFP)表达,计算能使 50% 细胞感染的稀释度,换算感染滴度。传统 “金标准”,但主观性强(依赖人工计数荧光细胞),检测周期长(3-5 天)。
流式细胞术(Flow Cytometry):若AAV携带荧光报告基因,可将其感染细胞后,通过流式细胞仪计数阳性细胞比例,结合稀释倍数计算感染滴度。客观性强(自动化计数),能同时分析细胞活力(排除细胞毒性干扰),已逐渐替代 TCID50 成为主流方法。
ICA(infectious center assay,复制中心测定法):采用AAV-rep和cap稳转细胞系作为AAV感染细胞,无需wtAAV共感染,避免或减少了实验环境中wtAAV污染,病毒感染后,会形成“复制中心”,通过原味杂交或免疫荧光来检测复制中心,在显微镜下计数阳性信号细胞。
活细胞分析: 活细胞成像可通过捕捉转导前后细胞的实时显微图像来评估转导滴度。该方法为转基因表达和细胞生理学提供了动态洞察。
3.2 目的序列表达水平检测
检测方法:从mRNA水平(qPCR)和蛋白水平(WB、ELISA、免疫染色)进行检测
mRNA 水平检测:验证基因转录效率
mRNA 水平直接反映目的序列的转录活性,核心检测目标是确认目的基因是否成功转录为功能性 mRNA,同时量化转录产物的含量,避免因启动子失效、序列突变导致转录受阻。
蛋白水平检测:验证基因翻译功能
蛋白水平是目的序列表达的最终功能体现,需同时确认蛋白的存在性、含量及活性,避免因密码子偏好性、翻译后修饰异常导致蛋白无功能或活性不足。
AAV 载体纯度直接影响临床安全性 —— 杂质(如聚集颗粒、宿主细胞残留)会引发免疫反应(如补体激活、抗体产生),空壳颗粒虽无毒性,但会竞争结合细胞表面受体,降低有效载体转导效率,同时由于病毒载量较高而增加免疫反应的风险。因此,纯度控制需重点关注 “衣壳纯度”“空壳率” 与 “聚集率” 三大指标,通过高分辨率分离技术精准量化。
衣壳纯度指 AAV 衣壳蛋白占总蛋白的比例,需排除生产残留的宿主细胞蛋白(HCP)、牛血清白蛋白(BSA,培养基成分)、蛋白酶(纯化用酶)等杂质蛋白 —— 这些杂质是主要免疫原性来源(如 HCP 会诱导抗 HCP 抗体产生)。
常用检测方法:SDS-PAGE、ELISA、LC-MS/MS
SDS-PAGE 电泳联合银染技术:除观察 VP1/VP2/VP3 条带外,需确认无杂带(杂质蛋白);若有微弱杂带,需通过 Western Blot 鉴定是否为 HCP,并通过 ELISA 定量。
酶联免疫吸附实验(ELISA):使用针对特定宿主细胞蛋白(HCPs, 如来自HEK293或Sf9细胞)或质粒编码蛋白(如Rep蛋白)的特异性抗体进行检测。
液相色谱-质谱联用(LC-MS/MS):目前最强大、最全面的非衣壳蛋白检测和鉴定技术,是深度表征的“黄金法则”。
在AAV载体生产过程中,除了产生所需全长目的基因的完整AAV载体,体系中还伴有空衣壳的病毒颗粒、填充了部分基因的病毒颗粒、基因组DNA过度装载的病毒颗粒、宿主DNA碎片、质粒DNA碎片和核酸酶等杂质的产生。
空壳颗粒是 AAV 生产不可避免的副产物(衣壳组装时未包裹基因组或包裹不完整基因组形成),表面结构与满壳颗粒一致,会竞争结合细胞受体降低转导效率,还可能激活免疫系统(如触发补体系统),是关键质量控制参数。
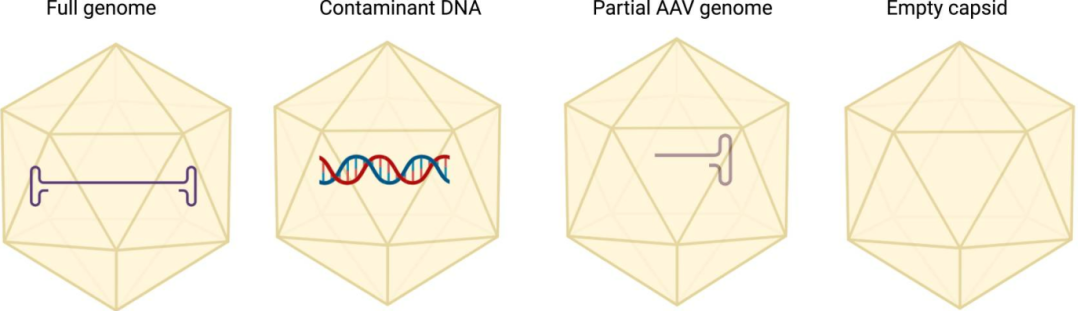
图1 在 AAV 生产批次中可以找到的各种 AAV 产品示意图(PMID: 39234444)
TEM(透射电子显微镜):直接观察衣壳群体,提供定性和半定量测量。
AUC(分析型超速离心):在强大的离心力场下,使空壳颗粒、部分填充壳、满壳颗粒以不同的速度沉降,形成不同的区带,通过光学系统(紫外或干涉)实时监测沉降过程,可以得到沉降系数分布图,从而精确定量空壳和完整壳的比例。当前空壳率检测 “金标准”,但检测周期长(24-48 小时)、成本高(仪器维护贵),仅适用于临床批、注册批等关键批次确认。
CDMS(电荷检测质谱):通过电喷雾电离将 AAV 颗粒转化为带电离子,结合离子阱质谱检测每个离子的电荷与质量,直接计算空壳颗粒与满壳颗粒电核质量比例,且能区分 “部分填充壳”(如包裹一半基因组的颗粒)。近年兴起的先进技术,已被 FDA 纳入 “先进检测方法” 名录,未来有望成为空壳率检测 “首选方法”。
IEC/AEX(离子交换色谱法):以阳离子交换树脂为固定相,基于空壳与满壳颗粒表面电荷差异(满壳因包裹带负电 DNA,表面电荷更低)实现分离,满壳颗粒保留时间长于空壳,通过峰面积比例计算空壳率。检测周期短(1-2 小时)、成本低,准确率≥85%,适合中试与商业化批次常规放行检测,但需控制样品缓冲液离子强度。
另一种关键杂质,衣壳聚集,会显著影响 AAV 的稳定性、安全性和传导效率。
常用检测方法:DLS、SEC
DLS(动态光散射):测量溶液中 AAV 颗粒的尺寸分布,在可见沉淀出现之前检测早期聚集。
SEC(尺寸排阻色谱法):分离并量化聚集的 AAV 颗粒与单体 AAV 颗粒,通常与多角度光散射(SEC-MALS)结合使用,以准确测定尺寸。
AAV 生产过程中会产生多种杂质,需建立针对性检测与控制策略。
宿主细胞残留杂质:包括宿主细胞 DNA(HCD)、宿主细胞蛋白(HCP)等。
工艺相关杂质:包括核酸酶残留、质粒残留等。
常用检测方法:ELISA、qPCR、HPLC
ELISA(酶联免疫吸附法):如基于固相酶联免疫吸附法,采用双抗体夹心的方式检测样品中HEK293来源的HCP残留量。
qPCR(实时荧光定量 PCR):可检测宿主细胞DNA残留、质粒DNA残留、宿主细胞残留DNA片段大小等
HPLC(高效液相色谱):可检测裂解剂残留、转染试剂残留等。
安全性是 AAV 质控的核心目标,需覆盖生产全流程及产品全生命周期。支原体、内毒素和外来因子等污染物会带来重大安全风险,必须经过严格测试。外来因子是细胞培养和载体生产过程中可能产生的意外病毒或微生物污染物。其中支原体污染会改变 AAV 效力和细胞活力; 内毒素(来自革兰氏阴性细菌的脂多糖)可引发严重的免疫反应。
无菌检测
通过直接接种法和膜过滤法,使用适宜的培养基检查需氧菌、厌氧菌和真菌。这是所有注射用生物制品的必检项目。
支原体检测方法
PCR:利用支原体16s rRNA和23s rRNA保守序列设计引物进行PCR和琼脂糖凝胶电泳。
成品培养基培养:基于培养的方法(如琼脂培养)通常用于检测。
指示细胞培养法:将样品与Vero等指示细胞共同培养,后用Hoechst 33258等DNA染料染色,在荧光显微镜下观察细胞周围或胞内是否有支原体DNA形成的荧光颗粒。此法更快(3-5天)。
细菌内毒素残留检测
鲎试剂吸光度测定:利用鲎试剂与内毒素反应过程中产生的凝固酶分解人工合成的显色基质,测定黄色的对硝基苯胺的生成量,进而检测内毒素含量,其是内毒素定量的黄金标准。
细菌检测
通过对细菌16S核糖体RNA(16S ribosomal RNA gene, 16S rRNA)基因特征核酸序列的PCR扩增及比对分析,实现细菌的分子生物学鉴定。
病毒污染检测
PCR和qPCR: 使用特异性引物和探针,扩增和检定特定外源病毒的核酸序列。
细胞病变效应和血吸附试验:将待测样品接种到多种指示细胞(如MRC-5, Vero)上,观察细胞病变,并进行血吸附试验(加入豚鼠红细胞),检测能凝集红细胞的病毒(如副流感病毒)。这是药典收录的经典方法。
动物接种法:将样品接种到乳鼠、成鼠和鸡胚等动物体内,观察动物是否发病或死亡。
AAV载体的关键质量属性(CQA)系统构成了其质量控制与批次放行的科学基础。从衣壳与基因组的身份鉴别,到基因组滴度、感染滴度等多层次效价评估,再到空壳率、聚集率及各类工艺杂质的精准控制,每一步都直接关联产品的疗效与安全。随着分析技术的不断进步(如ddPCR、CDMS、NGS等),AAV质控体系正向着更高分辨率、自动化及实时监控的方向发展。建立并持续完善一套科学、严谨且符合监管要求的CQA评价体系,是推动AAV基因治疗产品从实验室走向商业化成功的关键保障。
参考文献
【1】Kontogiannis T, Braybrook J, McElroy C, et al. Characterization of AAV vectors: A review of analytical techniques and critical quality attributes. Mol Ther Methods Clin Dev. 2024;32(3):101309. Published 2024 Jul 30. doi:10.1016/j.omtm.2024.101309
【2】Park MT, Verma A, Froelich CA, Motevalian SP. Process and quality considerations for recombinant adeno-associated virus manufacturing platforms. Trends Biotechnol. 2025;43(8):1921-1937. doi:10.1016/j.tibtech.2025.02.016
【3】国家药品监督管理局药品审评中心. 重组腺相关病毒载体类体内基因治疗产品临床试验申请药学研究与评价技术指导原则. 2024 年 01 月
【4】国际人用药品注册技术协调会(ICH).质量标准:生物技术产品及生物制品的检测方法和可接受标准. 1999 年 03 月

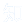


地址:-
地址:-
